Beyond Single Markers: How Systems Biology is Revolutionizing Biomarker Discovery and Precision Medicine
This article explores the paradigm shift from traditional reductionist biomarker approaches to holistic systems biology strategies in biomedical research and drug development.
Beyond Single Markers: How Systems Biology is Revolutionizing Biomarker Discovery and Precision Medicine
Abstract
This article explores the paradigm shift from traditional reductionist biomarker approaches to holistic systems biology strategies in biomedical research and drug development. It examines the foundational principles of both methodologies, detailing how systems biology integrates multi-omics data, computational modeling, and network analysis to decipher complex disease mechanisms. The content covers practical applications in areas from stem cell therapy to neurology and oncology, addresses key challenges in implementation, and provides a comparative validation of how this integrative framework enhances biomarker identification, patient stratification, and therapeutic development. Aimed at researchers and drug development professionals, this analysis synthesizes current evidence to illustrate how systems-level thinking is overcoming the limitations of single-target hypotheses for complex diseases.
From Isolated Parts to Interacting Networks: Core Principles of Reductionist vs. Systems Approaches
Table of Contents
- Philosophical and Methodological Foundations
- Comparative Analysis: Performance and Applications
- Experimental Protocols in Practice
- Visualizing the Workflows
- The Scientist's Toolkit: Essential Research Reagents
Philosophical and Methodological Foundations
The pursuit of biological knowledge and therapeutic breakthroughs is guided by two dominant paradigms: reductionism and systems holism. The reductionist approach, a long-standing cornerstone of biological research, operates on the principle that complex systems can be understood by isolating and studying their individual components, such as a single gene, protein, or pathway [1]. This methodology has been instrumental in identifying specific molecular players in disease. In contrast, systems biology is an interdisciplinary field that posits that the properties of a biological system cannot be fully understood by the study of its parts in isolation [1]. It argues that complexity arises from the dynamic networks of interactions between these components, and it applies computational and mathematical methods to study these complex interactions as integrated wholes [1].
The evolution of these fields is closely tied to technological advancements. Reductionist methods often rely on targeted assays, such as PCR for gene expression or ELISA for protein quantification, which focus on a single data type. Systems biology, however, is powered by high-throughput multi-omics technologies—including genomics, transcriptomics, proteomics, and metabolomics—that generate massive, multidimensional datasets [1] [2] [3]. The inherent complexity of human biological systems and multifactorial diseases like cancer and Alzheimer's has revealed the limitations of a purely reductionist, "single-target" approach, which often proves inadequate for achieving sufficient efficacy in the clinic [1]. This has driven the emergence of systems biology as a novel, innovative tool to tackle complex disease mechanisms and optimize drug discovery and development [1].
Comparative Analysis: Performance and Applications
The choice between reductionist and systems biology paradigms has profound implications for research outcomes, particularly in biomarker discovery and drug development. The table below summarizes a comparative analysis of the two approaches based on key performance indicators.
Table 1: Comparative Performance of Reductionist and Systems Biology Approaches
| Aspect | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Core Philosophy | Isolate and study single entities (e.g., a gene, protein) to understand the whole [1]. | Study the system as an integrated network of interacting components [1]. |
| Typical Data Type | Single-omics or targeted assays (e.g., PCR, ELISA) [2]. | Multi-omics (genomics, proteomics, metabolomics) and imaging data [2] [3]. |
| Handling of Complexity | Limited ability to capture multifaceted biological networks [2]. | Designed to address complexity and emergent properties of systems [1]. |
| Biomarker Discovery | Focus on single molecular features; faces challenges with reproducibility and predictive accuracy in complex diseases [2]. | Integrates diverse data to identify reliable, multi-component biomarker signatures; enables disease endotyping [2]. |
| Drug Development | "Single-target" drug development; less effective for complex diseases, leading to high clinical trial failure rates [1]. | Identifies combination therapies; matches right mechanism, dose, and patient population to increase probability of success [1]. |
| Key Strength | High precision for well-defined, single-factor problems; simpler experimental validation. | Superior for modeling complex, multifactorial diseases and predicting system-level responses [1]. |
| Primary Limitation | Inadequate for diseases driven by network dysregulation; higher risk of translational failure [1]. | Requires sophisticated computational infrastructure and expertise; challenges with model interpretability and uncertainty [2] [4]. |
Experimental Protocols in Practice
To illustrate these paradigms in action, below are generalized protocols for a typical biomarker discovery pipeline using each approach.
Protocol 1: Reductionist Approach for a Single-Protein Biomarker
This protocol aims to identify and validate a single protein biomarker, such as P-tau217 for Alzheimer's disease, from blood samples [5].
- Sample Collection and Processing: Collect blood plasma samples from clinically characterized cohorts (e.g., patients with cognitive impairment and healthy controls). Process blood to isolate plasma and aliquot for storage at -80°C.
- Targeted Assay (Simulated ELISA):
- Coating: Coat a 96-well plate with a capture antibody specific to the target protein (e.g., P-tau217).
- Blocking: Block remaining binding sites with a non-reactive protein (e.g., BSA).
- Sample Incubation: Add plasma samples and standards of known concentration to the wells. Incubate to allow the target antigen to bind the capture antibody.
- Detection Antibody Incubation: Add a detection antibody specific to a different epitope of the target protein. This antibody is conjugated to an enzyme (e.g., Horseradish Peroxidase).
- Signal Development: Add an enzyme substrate that produces a colorimetric or chemiluminescent signal proportional to the amount of target protein present.
- Data Acquisition: Measure the signal intensity using a plate reader.
- Data Analysis: Generate a standard curve from the known standards and calculate the concentration of the target protein in each unknown sample. Use statistical tests (e.g., t-test) to compare protein levels between patient and control groups.
Protocol 2: Systems Biology Approach for a Multi-Omics Biomarker Signature
This protocol leverages high-throughput technologies and machine learning to discover a composite biomarker signature from the same set of samples [1] [2] [3].
- Sample Collection and Multi-Omics Profiling:
- From a single aliquot of each plasma sample, perform parallel high-throughput molecular profiling:
- Genomics: Isolate DNA and perform whole-genome or exome sequencing to identify genetic variants.
- Transcriptomics: Isolate RNA from blood cells and perform RNA sequencing (RNA-seq) to quantify gene expression.
- Proteomics: Use mass spectrometry to quantify the levels of thousands of proteins.
- Metabolomics: Use mass spectrometry or NMR to profile small-molecule metabolites.
- From a single aliquot of each plasma sample, perform parallel high-throughput molecular profiling:
- Data Preprocessing and Integration:
- Quality Control: Process raw data from each platform using platform-specific pipelines (e.g., alignment for sequencing, peak identification for mass spec) to generate quantitative matrices.
- Normalization: Normalize data within each platform to correct for technical variance.
- Data Integration: Use computational methods to combine the different omics datasets into a unified data structure for each sample.
- Machine Learning-Based Biomarker Identification:
- Feature Selection: Apply feature selection algorithms (e.g., LASSO) to the integrated multi-omics data to identify a minimal set of genes, proteins, and metabolites that best predict the clinical outcome (e.g., disease state) [2].
- Model Training: Train a supervised machine learning model (e.g., Random Forest or Support Vector Machine) using the selected features on a training subset of the data [2].
- Model Validation: Test the trained model's performance on a held-out validation cohort to assess its predictive accuracy and generalizability.
- Systems-Level Validation (Optional): Place the identified biomarker signature into the context of known biological pathways (e.g., KEGG, Reactome) using pathway enrichment analysis to interpret the functional relevance of the findings.
Visualizing the Workflows
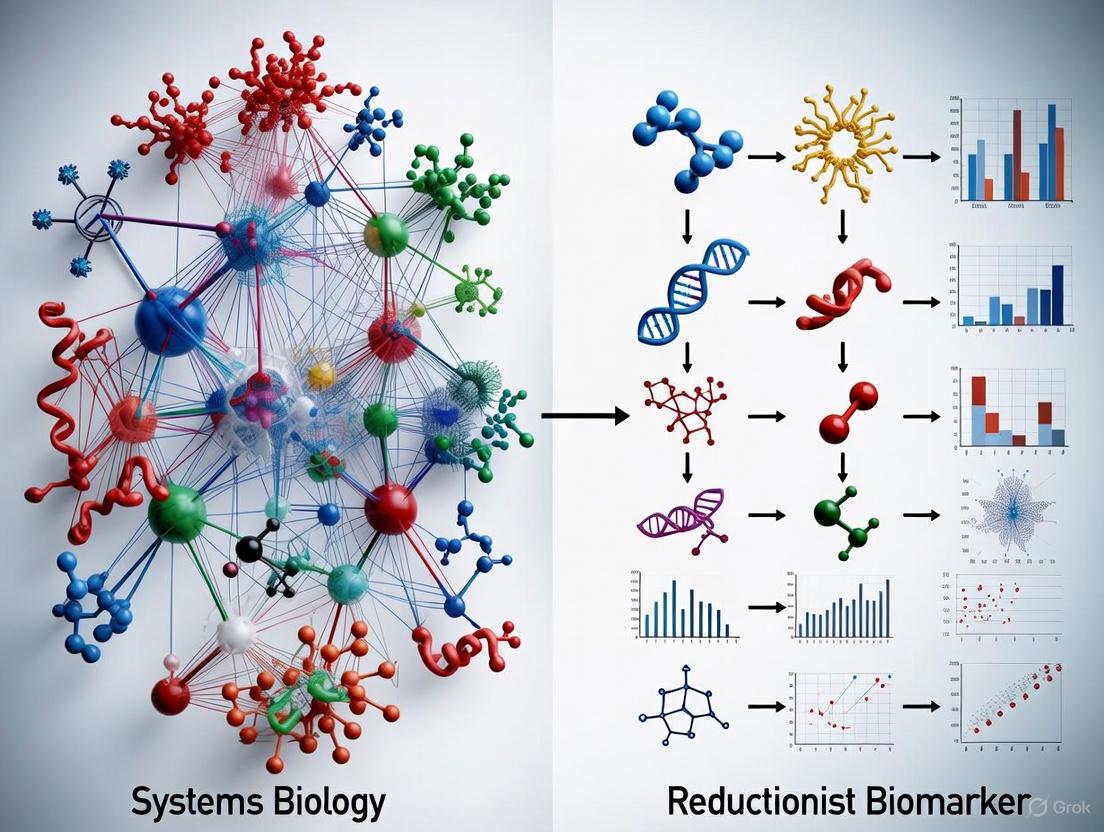
The fundamental difference in logic and workflow between the two paradigms can be visualized as a linear path versus an integrative network.
Reductionist Research Workflow
Systems Biology Research Workflow
The Scientist's Toolkit: Essential Research Reagents
The execution of these experimental protocols relies on a specific set of reagents and platforms. The following table details key solutions for both methodological paths.
Table 2: Essential Research Reagent Solutions for Biomarker Discovery
| Reagent / Platform | Function | Commonly Used In |
|---|---|---|
| ELISA Kits | Quantifies the concentration of a specific target protein in a solution using enzyme-linked antibodies. | Reductionist Approach [5] |
| PCR & qRT-PCR Assays | Amplifies and quantifies specific DNA or RNA sequences from a sample. | Reductionist Approach |
| Next-Generation Sequencing (NGS) | High-throughput technology for determining the sequence of DNA (genomics) or RNA (transcriptomics) [2]. | Systems Biology Approach |
| Mass Spectrometer | High-sensitivity instrument that identifies and quantifies proteins (proteomics) and metabolites (metabolomics) in a sample [1] [2]. | Systems Biology Approach |
| Spatial Biology Platforms | Enables in-situ analysis of gene expression (spatial transcriptomics) and protein multiplexing, preserving the tissue's spatial architecture [6] [3]. | Systems Biology Approach |
| AI/ML Software (e.g., R, Python scikit-learn) | Provides algorithms for integrating multi-omics data, performing feature selection, and training predictive models [2] [7]. | Systems Biology Approach |
| Human Organoids | 3D cell cultures that mimic human tissue architecture and function, used for functional validation of biomarkers in a human-relevant context [3]. | Both (Advanced Validation) |
The field of biomarker discovery has been fundamentally shaped by a reductionist approach that dominated biological research for decades. This paradigm operates on the principle that complex biological systems are best understood by breaking them down into their constituent parts and studying each component in isolation. In the context of biomarkers, this translated to a research model focused on identifying single, discrete biological indicators—a "one mutation, one target, one test" methodology [6]. This single-target framework produced remarkable successes, particularly in the late 20th century, establishing biomarkers as valuable tools for understanding disease mechanisms, identifying drug targets, and monitoring therapeutic responses [7].
The historical preference for single-target discovery was not merely philosophical but largely technology-driven. Research teams were constrained by the tools available: low-throughput assays, limited computational power, and biochemical methods that excelled at measuring individual analytes rather than complex molecular networks. These methods included PCR for specific genetic mutations, ELISA for individual protein biomarkers, and immunohistochemistry for protein expression patterns in tissues [8] [3]. The success of this approach is evidenced by foundational biomarkers such as HER2 for breast cancer stratification and PSA for prostate cancer detection, which revolutionized diagnostic and treatment paradigms in their respective fields [9].
However, as biomedical research has advanced, the inherent limitations of this single-target approach have become increasingly apparent. Complex diseases like cancer, autoimmune disorders, and neurological conditions seldom arise from dysfunction in a single biological pathway but rather emerge from dysregulated networks of molecular interactions [10] [11]. This recognition, coupled with technological advances enabling measurement of thousands of molecular features simultaneously, has prompted a fundamental shift toward systems biology approaches that embrace rather than reduce biological complexity [8] [10].
Historical Successes of Single-Target Biomarker Discovery
Foundational Discoveries and Clinical Impact
The single-target biomarker approach has yielded numerous critical discoveries that formed the foundation of modern diagnostic medicine. These biomarkers provided the first objective measures for disease detection, risk stratification, and treatment monitoring, moving medical practice beyond reliance on subjective symptoms alone. The most impactful successes came from oncology, where biomarkers like carcinoembryonic antigen (CEA) and alpha-fetoprotein (AFP) established in the 1970s provided the first measurable indicators of tumor presence and burden [9]. These discoveries demonstrated that molecular signatures could offer clinically valuable information about disease state, paving the way for more personalized approaches to cancer management.
The paradigm further evolved with the development of predictive biomarkers that could forecast response to specific therapies. The landmark discovery of HER2 overexpression in a subset of breast cancers and its correlation with dramatic response to HER2-targeted therapies like trastuzumab exemplified the power of single-target biomarkers to guide therapeutic decisions [9]. This "one drug, one biomarker" model became the gold standard for drug development in oncology and beyond, enabling more precise targeting of treatments to patients most likely to benefit. Similarly, EGFR mutations in lung cancer became crucial predictors of response to tyrosine kinase inhibitors, transforming treatment outcomes for specific molecular subsets of patients [9].
Methodological Contributions and Diagnostic Frameworks
The single-target approach established essential methodological frameworks that continue to underpin biomarker research. It developed standardized assay validation protocols, reference standards, and analytical performance metrics that ensured reliability and reproducibility in clinical measurements [7]. The rigorous validation pathways established for these biomarkers created templates for regulatory approval processes, with clear evidence requirements for analytical validity, clinical validity, and clinical utility [6].
The technological legacy of this era is equally significant. Single-target discovery drove innovations in assay sensitivity, specificity, and reproducibility across various testing platforms. It established core laboratory methodologies including PCR-based genotyping, immunoassay development, and chromatographic techniques for measuring small molecules [8]. These technical advances created the foundation upon which modern multiplexed assays would later be built. The clinical diagnostic paradigms established through single-target biomarkers—including companion diagnostics, laboratory-developed tests, and standardized reporting frameworks—created the infrastructure necessary for integrating molecular information into routine clinical decision-making [7] [9].
Table 1: Historic Single-Target Biomarkers and Their Clinical Impact
| Biomarker | Disease Context | Clinical Application | Impact |
|---|---|---|---|
| HER2 | Breast Cancer | Predicts response to trastuzumab and other HER2-targeted therapies | Established paradigm for targeted therapy in molecularly-defined subsets |
| EGFR mutations | Non-Small Cell Lung Cancer | Predicts response to EGFR tyrosine kinase inhibitors | Transformed treatment landscape for lung cancer, improving outcomes in molecularly selected patients |
| BRCA1/2 mutations | Hereditary Breast and Ovarian Cancer | Risk assessment and prevention strategies | Enabled prophylactic interventions and personalized screening protocols |
| PD-L1 expression | Multiple Cancers | Guides immunotherapy decisions | Identifies patients most likely to benefit from immune checkpoint inhibitors, though with limitations |
| KRAS mutations | Colorectal Cancer | Predicts resistance to anti-EGFR therapy | Prevents ineffective treatments and spares patients from unnecessary toxicity |
Limitations of the Single-Target Approach
Biological Complexity and Disease Heterogeneity
The fundamental limitation of single-target biomarker discovery lies in its inability to capture the multidimensional nature of most disease processes. Complex diseases arise from dysregulated networks of molecular interactions rather than isolated defects in single pathways [10] [11]. This biological reality means that measuring individual components often provides an incomplete picture of disease pathogenesis, progression, or therapeutic responsiveness. The reductionist approach inherently oversimplifies diseases that are themselves complex adaptive systems with emergent properties not predictable from individual components [10].
This limitation manifests clinically as inconsistent predictive value across diverse patient populations. For example, while PD-L1 expression helps guide immunotherapy decisions, response rates vary significantly even among patients with high PD-L1 expression, indicating that this single parameter cannot fully capture the complexity of tumor-immune interactions [9]. Similarly, the heterogeneity of tumors means that biopsies from different regions of the same tumor may show different biomarker expression patterns, leading to sampling errors and false negatives when relying on single-target measurements [3]. Spatial biology techniques have revealed that biomarker distribution patterns within tissues often carry crucial clinical information that is lost when simply measuring presence or absence [3].
Methodological and Technological Constraints
The single-target approach suffers from several methodological limitations that restrict its clinical utility. The "one biomarker at a time" discovery process is inherently inefficient, requiring separate development and validation pathways for each candidate biomarker [12]. This linear model significantly delays the translation of discoveries into clinical practice and contributes to the high failure rate of biomarker candidates, with only 0-2 new protein biomarkers achieving FDA approval per year across all diseases [12].
The statistical challenges are equally formidable. Single-target biomarkers often demonstrate inadequate sensitivity or specificity when applied broadly, leading to both false positives and false negatives with significant clinical consequences [12]. The "small n, large p" problem—where the number of potential features (genes, proteins, etc.) far exceeds the number of patient samples—makes it statistically difficult to identify truly meaningful signals without sophisticated multivariate analytical approaches [12]. Furthermore, the snapshot nature of most single-target measurements fails to capture the dynamic nature of disease processes and treatment responses, providing limited information about disease trajectory or evolving therapeutic resistance [12] [13].
Table 2: Limitations of Single-Target Biomarker Approaches
| Limitation Category | Specific Challenges | Clinical Consequences |
|---|---|---|
| Biological Complexity | Inability to capture pathway interactions and network dynamics | Incomplete understanding of disease mechanisms and compensatory pathways |
| Disease Heterogeneity | Tumor heterogeneity and spatial variation in biomarker expression | Sampling errors, false negatives, and incomplete prognostic information |
| Analytical Performance | Inadequate sensitivity/specificity for complex diseases | Misdiagnosis, missed diagnoses, and incorrect treatment assignments |
| Technological Constraints | Static measurements that miss dynamic disease processes | Inability to monitor real-time treatment response and evolving resistance mechanisms |
| Statistical Challenges | High false discovery rates with multiple hypothesis testing | Many biomarker candidates fail validation, wasting resources and delaying progress |
The Systems Biology Alternative: A Comparative Framework
Philosophical and Methodological Differences
Systems biology represents a paradigm shift from the reductionist approach, founded on the principle that biological systems must be understood as integrated networks rather than collections of isolated components [10]. Where reductionism seeks to simplify complexity by studying parts in isolation, systems biology embraces complexity by examining interactions and emergent properties of whole systems [10] [11]. This philosophical difference manifests methodologically through the use of high-throughput technologies, computational modeling, and network analysis to capture the multidimensional nature of biological processes [10].
The contrast between these approaches is evident in their respective workflows. While single-target discovery follows a linear path from hypothesis to validation of individual candidates, systems biology employs iterative cycles of computational modeling and experimental validation that continuously refine understanding of the entire system [10]. Rather than testing predefined hypotheses about specific molecules, systems approaches often begin with agnostic data collection across multiple biological layers (genomics, transcriptomics, proteomics, etc.), using computational methods to identify patterns that emerge from the data itself [8] [9]. This data-driven discovery process can reveal novel relationships that would not have been hypothesized through traditional reductionist frameworks.
Technological and Analytical Advancements
The systems approach is enabled by technological advances that allow comprehensive molecular profiling at multiple levels. Multi-omics platforms simultaneously capture data from genomics, transcriptomics, proteomics, and metabolomics, providing a layered view of biological systems that captures their inherent complexity [8] [6] [13]. Spatial biology techniques preserve the architectural context of biomarkers within tissues, revealing how cellular organization and proximity influences function—information completely lost in single-target approaches that homogenize tissues [3]. Single-cell analysis technologies resolve cellular heterogeneity that is averaged out in bulk measurements, identifying rare cell populations that may drive disease progression or treatment resistance [13].
The analytical framework of systems biology represents an equally significant advancement. Network analysis using tools like Cytoscape maps molecular interactions to identify key regulatory nodes and pathways [10] [11]. Artificial intelligence and machine learning algorithms detect complex, non-linear patterns in high-dimensional data that escape conventional statistical methods [8] [7] [9]. These computational approaches can integrate multimodal data—combining molecular profiles with clinical information, medical images, and real-world evidence—to generate more comprehensive biomarkers that better reflect biological reality [7] [9].
Diagram 1: Comparison of reductionist and systems biology approaches to biomarker discovery shows fundamental differences in process flow and philosophy.
Comparative Experimental Data: Single-Target vs. Systems Approaches
Direct Methodological Comparisons
The contrast between single-target and systems approaches becomes evident when examining their application to specific disease contexts. In inflammatory bowel disease (IBD), traditional single-target studies focused on individual cytokines (e.g., TNF, IL6) or genetic variants (e.g., NOD2) provided limited insights into the complex pathophysiology distinguishing Crohn's disease from ulcerative colitis [11]. When researchers applied a systems biology approach—constructing causal biological network models that integrated multiple signaling pathways—they identified distinct network perturbation patterns between these related conditions [11]. The systems model revealed that in the "intestinal permeability" network, programmed cell death factors were downregulated in Crohn's disease but upregulated in ulcerative colitis, while in the "wound healing" network, pro-healing factors showed opposite regulation patterns between the two diseases [11].
Similar advantages emerge in oncology. While single-target biomarkers like HER2 or EGFR mutations provide valuable but limited information, AI-powered analysis of multi-omics data can identify composite biomarker signatures with superior predictive power [7] [9]. For example, in colorectal cancer, deep learning analysis of standard histopathology images identified prognostic patterns that outperformed established molecular and morphological markers [7]. These systems-level biomarkers capture the complex interactions between tumor cells, immune infiltrates, and stromal components that single-target approaches cannot represent [3] [9].
Performance Metrics and Validation Outcomes
Quantitative comparisons demonstrate the enhanced performance of systems approaches across multiple metrics. Single-target biomarkers typically show moderate accuracy (often 70-80% sensitivity/specificity) for complex endpoints, reflecting their inherent limitation of reducing multidimensional biology to univariate measurements [12] [9]. In contrast, multimodal AI biomarkers that integrate genomic, imaging, and clinical data have demonstrated 15% improvement in survival risk prediction in phase 3 clinical trials compared to traditional approaches [9].
The validation outcomes further highlight these differences. The development pathway for single-target biomarkers is characterized by high attrition rates, with the "verification tar pit" consuming up to $2 million and over a year per candidate, often ending in failure [12]. Systems approaches that identify biomarker panels or signatures face different validation challenges but demonstrate better generalizability across diverse populations when properly developed [8] [12]. The validation of single-target biomarkers typically requires thousands of samples to achieve adequate statistical power, while systems approaches using machine learning may require even larger datasets but can extract more information from each sample [12] [9].
Table 3: Quantitative Comparison of Single-Target vs. Systems Biology Approaches
| Performance Metric | Single-Target Approach | Systems Biology Approach |
|---|---|---|
| Development Timeline | Years for single candidates | Months for signature discovery |
| Attrition Rate | Very high (>95% failure) | High but with more validated outputs per study |
| Predictive Accuracy for Complex Diseases | Moderate (typically 70-80% AUC) | Higher (typically 80-90% AUC for best validated models) |
| Biological Coverage | Narrow (single pathway) | Comprehensive (multiple interacting pathways) |
| Handling of Heterogeneity | Poor (misses spatial and temporal variation) | Better (can incorporate spatial context and dynamics) |
| Clinical Implementation | Simpler regulatory path | More complex validation requirements |
| Cost per Candidate | Up to $2M verification cost | Higher initial investment but more information per study |
The Scientist's Toolkit: Essential Research Reagents and Platforms
Core Technologies for Biomarker Discovery
Transitioning from single-target to systems biomarker discovery requires both conceptual shifts and adoption of new technological platforms. The modern biomarker discovery toolkit encompasses technologies that enable comprehensive molecular profiling, spatial contextualization, and computational integration of diverse data types [6] [3]. Multi-omics profiling platforms form the foundation, with next-generation sequencing providing genomic and transcriptomic data, mass spectrometry enabling proteomic and metabolomic measurements, and emerging technologies like spatial transcriptomics capturing molecular information within architectural context [6] [3]. For example, Element Biosciences' AVITI24 system combines sequencing with cell profiling to simultaneously capture RNA, protein, and morphological data, while 10x Genomics platforms enable millions of cells to be analyzed at once [6].
Advanced model systems constitute another critical component of the modern toolkit. Organoid cultures recapitulate the complex architecture and functions of human tissues more faithfully than traditional 2D cell lines, making them valuable for functional biomarker screening and target validation [3]. Humanized mouse models incorporate human immune system components, enabling studies of human-specific tumor-immune interactions and immunotherapy response biomarkers [3]. When used in conjunction with multi-omics technologies, these advanced models enhance the translational relevance of biomarker discoveries by better mimicking human biology and disease processes [3].
The computational infrastructure for systems biomarker discovery represents perhaps the most significant departure from traditional approaches. AI and machine learning platforms are essential for analyzing the high-dimensional data generated by multi-omics technologies [7] [9]. These include deep learning algorithms for pattern recognition in complex datasets, natural language processing for extracting insights from clinical narratives, and explainable AI methods that make computational predictions interpretable to clinicians [7] [9]. Open-source resources like the Digital Biomarker Discovery Pipeline (DBDP) provide standardized toolkits and reference methods that promote reproducibility and collaboration [12].
Data management and integration systems form the backbone of modern biomarker discovery operations. Federated learning approaches enable analysis across distributed datasets without moving sensitive patient data, addressing privacy concerns while maximizing available information [9]. Cloud computing platforms provide the scalable computational resources needed for large-scale multi-omics analyses, while laboratory information management systems (LIMS) and electronic data capture systems maintain sample integrity and data quality throughout the discovery pipeline [6] [12]. Together, these technologies create an integrated ecosystem that supports the complex, data-intensive workflow of systems biomarker discovery from initial measurement through clinical validation.
Diagram 2: Modern systems biology workflow for biomarker discovery integrates multiple data types and emphasizes computational analysis.
Table 4: Essential Research Reagent Solutions for Modern Biomarker Discovery
| Technology Category | Specific Tools/Platforms | Primary Function | Key Applications |
|---|---|---|---|
| Multi-Omics Profiling | Next-generation sequencing, Mass spectrometry, Microarrays | Comprehensive molecular measurement across biological layers | Biomarker identification, Pathway analysis, Molecular subtyping |
| Spatial Biology | Multiplex immunohistochemistry, Spatial transcriptomics, Imaging mass cytometry | Preserve architectural context of biomarkers within tissues | Tumor microenvironment characterization, Cellular interaction mapping |
| Single-Cell Technologies | Single-cell RNA sequencing, CyTOF, Cellular indexing | Resolve cellular heterogeneity masked in bulk measurements | Rare cell population identification, Cellular trajectory reconstruction |
| Advanced Model Systems | Organoids, Humanized mouse models, 3D culture systems | Better mimic human biology and disease processes | Functional biomarker validation, Therapeutic response prediction |
| Computational Platforms | AI/ML algorithms, Network analysis tools, Cloud computing | Analyze high-dimensional data and identify complex patterns | Predictive model development, Biomarker signature discovery |
The historical context of single-target biomarker discovery reveals both remarkable achievements and inherent limitations. The reductionist approach produced foundational biomarkers that transformed diagnostic and therapeutic paradigms in multiple disease areas, particularly oncology, while establishing methodological standards and regulatory pathways that continue to guide biomarker development [7] [9]. Its limitations in addressing complex, multifactorial diseases reflect not scientific failure but rather the boundary of what was technologically and conceptually possible during its ascendancy [10].
The ongoing shift toward systems biology does not render single-target approaches obsolete but rather recontextualizes them within a more comprehensive framework [8] [10]. Single-target biomarkers continue to provide clinical value in specific contexts where diseases are driven by discrete molecular events. However, for most complex diseases, the future lies in integrated approaches that combine the methodological rigor of reductionism with the comprehensive perspective of systems biology [11] [9]. This synthesis leverages technological advances in multi-omics profiling, spatial biology, and computational analysis to develop biomarker signatures that better reflect the multidimensional nature of health and disease [6] [13].
The most productive path forward recognizes that these approaches are complementary rather than contradictory. Single-target biomarkers provide focused insights with clear clinical actionability, while systems approaches capture the complexity that single targets miss [10] [9]. The future of biomarker discovery lies not in choosing between these paradigms but in developing frameworks that integrate their respective strengths, leveraging historical wisdom while embracing technological innovation to advance personalized medicine [8] [13].
Systems biology represents a fundamental paradigm shift in biological research, moving from the traditional reductionist approach to a holistic perspective that seeks to understand how biological components interact to form functional systems. Where reductionism focuses on isolating and studying individual biological parts—single genes, proteins, or pathways—systems biology investigates the complex networks of interactions that give rise to emergent behaviors not predictable from individual components alone [14] [15]. This philosophical shift began in the early 20th century as scientists recognized the limitations of purely mechanistic approaches that interpreted organisms as simple clockwork-like machines [14].
The foundational revolution in systems thinking accelerated with Roger Williams' groundbreaking 1956 work, which compiled extensive evidence of molecular, physiological, and anatomical individuality in animals [14]. Williams demonstrated that normal, healthy individuals exhibit enormous variation—often 20 to 50-fold differences in biochemical, hormonal, and physiological parameters—revealing that the "average individual" is a statistical abstraction rather than a biological reality [14]. This evidence directly contradicted strict mechanistic views and revealed that living systems possess robust compensation mechanisms that maintain function despite significant molecular variation, a core systems property [14].
Table 1: Fundamental Contrasts Between Reductionist and Systems Biology Approaches
| Aspect | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Primary Focus | Isolated components | Networks and interactions |
| Core Philosophy | Breaking down systems into constituent parts | Understanding emergence from system interactions |
| Methodology | Studies elements in isolation | Studies systems as integrated wholes |
| Variability Treatment | Often considered noise | Recognized as biologically significant |
| Modeling Approach | Linear causality | Nonlinear, dynamic networks |
| Experimental Design | Controlled, single-variable | Multi-parameter, high-throughput |
Core Principles of Systems Biology
Holism and Emergent Properties
The principle of holism constitutes the foundational tenet of systems biology, positing that "the whole is something over and above its parts and not just the sum of them all" [14]. This Aristotelian concept, revitalized in modern systems science, emphasizes that biological systems exhibit emergent properties—unique characteristics possessed only by the whole system and not shared to any great degree by individual components in isolation [14] [15]. These emergent behaviors arise from the complex, dynamic interactions between system components and cannot be predicted by studying individual elements alone [16].
Living systems are characterized by their hierarchical organization, with systems nested within systems across multiple scales of complexity [14]. This hierarchical structure ranges from molecular networks and cellular systems to tissues, organs, organisms, and ecosystems. At each level, new properties emerge that are not present at lower levels, requiring specific approaches to study and understand these system-level behaviors [14]. The systems perspective recognizes that the structure of an entire system actually orchestrates and constrains the behavior of its component parts, creating downward causation effects that reductionist approaches cannot capture [14].
Networks and Interconnectivity
Biological networks represent the architectural framework through which emergent properties manifest in living systems. Systems biology represents biological relationships as interconnected networks where nodes symbolize system components (genes, proteins, metabolites) and connecting links represent interactions or reactions [10]. These networks can be constructed through various approaches: (1) de novo from direct experimental interactions; (2) by applying known interactions to experimental data using specialized software; or (3) through reverse engineering approaches that infer network structures from system behavior [10].
The interconnectivity within biological networks means that changes to one component inevitably influence others, often through complex feedback loops that can be either positive (amplifying changes) or negative (stabilizing systems) [16]. This network perspective reveals that biological functions are rarely regulated by single molecules but rather emerge from the coordinated interactions of multiple system components [10]. Understanding the network topology—the specific patterns of connections—becomes essential for identifying key regulatory points and understanding system dynamics and robustness [17] [16].
Diagram 1: Conceptual Framework of Systems Biology
Integration of Multi-Scale Data
Integration represents the methodological cornerstone of systems biology, enabling the synthesis of information across multiple biological levels and scales [15] [16]. This integrative approach combines diverse data types—genomic, transcriptomic, proteomic, metabolomic, and clinical—to construct comprehensive models of biological systems [17] [15]. The emergence of multi-omics technologies has transformed systems biology by providing extensive datasets that cover different biological layers, enabling a more profound comprehension of biological processes and interactions [15].
The integration process follows a cyclical framework of theory, computational modeling, hypothesis generation, experimental validation, and model refinement [15]. This iterative cycle accelerates discovery and enhances the reliability of predictions [18]. Successful integration requires sophisticated computational tools and methods for data integration and mining, including network analysis, machine learning, and pathway enrichment approaches [15] [16]. These methodologies enable researchers to extract meaningful patterns and insights from integrated datasets, moving beyond simple correlation to establish causal relationships within biological systems [10] [11].
Methodological Framework: The Systems Biology Toolkit
Computational and Modeling Approaches
Systems biology employs both top-down and bottom-up modeling strategies to understand biological complexity [15]. The top-down approach begins with system-level observational data, typically from high-throughput 'omics' technologies, and works downward to identify molecular interaction networks and generate hypotheses about regulatory mechanisms [15]. In contrast, the bottom-up approach starts from detailed mechanistic knowledge of individual components and their interactions, building upward to reconstruct system behavior from first principles [15].
Table 2: Computational Modeling Methods in Systems Biology
| Model Type | Key Features | Typical Applications |
|---|---|---|
| Ordinary Differential Equations (ODE) | Captures continuous dynamics of molecular interactions | Signaling pathways, metabolic networks |
| Boolean Networks | Simplified logical (ON/OFF) representation of component states | Gene regulatory networks, cellular fate decisions |
| Agent-Based Models | Simulates behaviors of individual entities and their interactions | Cellular populations, tissue organization |
| Network Models | Graph-based representation of component relationships | Protein-protein interaction maps, disease mechanism analysis |
| Multi-Scale Models | Integrates processes across different temporal and spatial scales | Organ-level physiology, host-pathogen interactions |
The bottom-up approach is particularly valuable in pharmaceutical applications, as it facilitates the translation of drug-specific in vitro findings to the in vivo human context [15]. This includes predicting drug exposure through physiologically based pharmacokinetic (PBPK) modeling and translating in vitro data on drug-ion channel interactions to physiological effects [15]. The separation of drug-specific, system-specific, and trial design parameters enables predictions of exposure-response relationships that account for inter- and intra-individual variability, making this approach particularly valuable for population-level drug effect assessments [15].
Experimental and Analytical Technologies
Modern systems biology relies on high-throughput technologies that enable the simultaneous measurement of thousands of system components [15] [16]. These technologies include next-generation sequencing for genomic characterization, mass spectrometry for proteomic and metabolomic profiling, and advanced imaging techniques for spatial and temporal analysis of biological systems [16]. The massive datasets generated by these technologies necessitate sophisticated computational infrastructure and bioinformatic tools for data management, processing, and analysis [10].
Network analysis represents a core analytical approach in systems biology, leveraging mathematical tools from Graph Theory to identify key regulatory nodes, network motifs, and functional modules within biological systems [10]. Software platforms like Cytoscape provide versatile environments for complex network visualization and analysis [10] [11]. The emerging integration of machine learning and artificial intelligence approaches further enhances the ability to detect hidden patterns in multi-omics data and predict system behaviors under different conditions [19] [18].
Diagram 2: Systems Biology Research Workflow
Comparative Analysis: Systems Biology vs. Reductionist Biomarker Approaches
Philosophical and Methodological Differences
The fundamental distinction between systems biology and reductionist biomarker approaches lies in their treatment of biological complexity. While reductionist methods typically seek to minimize complexity through controlled experiments that isolate single variables, systems biology embraces complexity by simultaneously measuring multiple system components and analyzing their interactions [15]. Reductionist approaches have proven highly successful in identifying individual biological components and their specific functions but offer limited capacity for understanding how system properties emerge from interactions [15].
Reductionist biomarker strategies typically focus on identifying single molecules or linear pathways as diagnostic or therapeutic indicators [10]. In contrast, systems biology recognizes that most biological features are determined by complex interactions among multiple system components, and therefore focuses on identifying biomodules—groups of interacting molecules that regulate discrete functions—and their interrelationships within larger networks [10]. This network perspective enables a more comprehensive understanding of disease mechanisms and treatment responses that cannot be captured by single biomarkers alone.
Practical Applications in Drug Development
The application of systems biology in pharmaceutical research has demonstrated significant advantages over traditional reductionist approaches, particularly for complex diseases involving multiple interacting pathways [11] [18]. Quantitative Systems Pharmacology (QSP) has emerged as a powerful application of systems biology in drug development, leveraging comprehensive biological models to simulate drug behaviors, predict patient responses, and optimize development strategies [20]. QSP approaches enable more informed decisions in drug discovery, potentially reducing development costs and bringing safer, more effective therapies to patients faster [20].
Table 3: Comparison of Applications in Inflammatory Bowel Disease Research
| Research Aspect | Reductionist Biomarker Approach | Systems Biology Approach |
|---|---|---|
| Barrier Function Analysis | Focuses on single tight junction proteins | Models integrated programmed cell death and tight junction networks |
| Inflammatory Response | Measures individual cytokines (e.g., TNF, IL6) | Captures PPARG, IL6, and IFN pathway interactions |
| Disease Differentiation | Relies on single discriminatory markers | Identifies distinct network perturbation patterns for CD vs. UC |
| Therapeutic Targeting | Targets single pathways | Identifies central network nodes and combination strategies |
| Personalization | Limited by single-molecule variability | Accounts for compensatory mechanisms within networks |
A concrete example of the systems approach can be found in Inflammatory Bowel Disease (IBD) research, where causal biological network models have been developed to represent signaling pathways contributing to Crohn's disease and ulcerative colitis [11]. These models integrate scientific knowledge using Biological Expression Language (BEL) to create computable network models that capture complex relationships between biological entities [11]. When scored with transcriptomic data from diseased tissues, these network models reveal distinct perturbation patterns between different IBD forms, providing mechanistic insights that single biomarker approaches cannot deliver [11].
Case Study: Network Analysis in Inflammatory Bowel Disease
Experimental Protocol and Workflow
The systems biology approach to IBD research exemplifies the power of network-based analysis for understanding complex disease mechanisms [11]. The research follows a structured workflow beginning with comprehensive literature curation to identify known signaling pathways involved in barrier defence, inflammatory processes, and wound healing in IBD [11]. This knowledge is formalized using Biological Expression Language (BEL), which converts relationships between biomolecules into cause-and-effect statements using controlled vocabularies that facilitate computational analysis [11].
Each BEL statement consists of a source, relationship, and target, where biological entities are defined by specific functions (RNA abundances, protein abundances, protein activities, etc.) and referenced using standard namespaces [11]. Contextual details including species, cell type, and disease state are captured as annotations with each statement [11]. The curated BEL statements are then compiled into network models using the OpenBEL framework and reviewed using Cytoscape to identify gaps and ensure completeness [11]. These computable network models enable quantitative analysis of transcriptomic data from diseased tissues, providing insights into network perturbations associated with specific disease states [11].
Key Findings and Comparative Insights
Application of this systems biology approach to IBD revealed distinct network perturbation patterns that differentiate Crohn's disease from ulcerative colitis [11]. In the "intestinal permeability" model, programmed cell death factors were downregulated in Crohn's disease but upregulated in ulcerative colitis [11]. The "inflammation" model highlighted PPARG, IL6, and IFN-associated pathways as prominent regulatory factors in both diseases, but with distinct interaction patterns [11]. Most strikingly, in the "wound healing" model, factors promoting wound healing were upregulated in Crohn's disease but downregulated in ulcerative colitis, providing mechanistic insights into their different clinical presentations and progression patterns [11].
These findings demonstrate how systems biology approaches can capture complex, multidimensional differences between related disease states that reductionist biomarker approaches typically miss. By analyzing network-wide perturbation patterns rather than individual molecule changes, systems biology provides a more comprehensive understanding of disease mechanisms and potential therapeutic interventions [11].
Essential Research Reagents and Computational Tools
The implementation of systems biology research requires specialized reagents and computational resources that enable comprehensive system characterization and modeling. The following table details key solutions essential for conducting systems biology investigations, particularly those focused on network analysis and multi-omics integration.
Table 4: Essential Research Reagent Solutions for Systems Biology
| Reagent/Tool | Primary Function | Application Example |
|---|---|---|
| OpenBEL Framework | Compiles biological relationships into computable network models | Formalizing causal relationships in IBD pathway models [11] |
| Cytoscape | Network visualization and analysis | Reviewing and analyzing biological network models [10] [11] |
| Ingenuity Pathway Analysis | Known interaction mapping from experimental data | Building biological networks from gene lists [10] |
| String Database | Protein-protein interaction data source | Constructing interaction networks from proteomic data [10] |
| Multi-omics Platforms | Simultaneous measurement of multiple biological layers | Integrating genomic, transcriptomic, proteomic data [15] [16] |
| High-Throughput Sequencers | Comprehensive molecular profiling | Generating genome-wide transcriptomic data [16] |
| Mass Spectrometers | Proteomic and metabolomic characterization | Quantitative measurement of protein abundances [10] |
Systems biology represents more than just a collection of computational techniques—it constitutes a fundamental philosophical shift in how we approach biological complexity [14] [15]. By focusing on networks, emergent properties, and integration, systems biology provides a powerful framework for understanding biological systems in their full complexity, overcoming limitations of traditional reductionist approaches that necessarily isolate components from their physiological context [15]. The core tenets of systems biology—holism, interconnectivity, emergence, and dynamic integration—provide a more accurate representation of biological reality, where function arises from the coordinated interactions of multiple components across different scales of organization [14] [16].
The comparative analysis between systems biology and reductionist biomarker approaches reveals that these perspectives are not mutually exclusive but rather complementary [14]. Reductionist approaches excel at identifying components and their specific functions, while systems biology explains why these components are organized as they are and how their interactions give rise to system-level behaviors [14]. The most powerful research strategies integrate both approaches, using reductionist methods to characterize individual components and systems approaches to understand their functional integration [14].
As systems biology continues to evolve, its impact on therapeutic innovation and personalized medicine continues to grow [20] [18]. By providing holistic insights into disease mechanisms and guiding rational intervention strategies, systems biology represents an essential tool for advancing the next generation of therapies [18]. It bridges the critical gap between data generation and clinical decision-making, ensuring that the vast amounts of biological information generated by modern technologies are translated into meaningful therapeutic outcomes for patients [18]. The continued development of educational programs [20] and collaborative industry-academia partnerships [20] will be essential for training the next generation of scientists capable of leveraging these powerful approaches to address the complex biological challenges of the future.
For the past half-century, epidemiology and disease research have been dominated by a reductionist paradigm focused on isolating single causes of disease states [21]. This approach, rooted in Koch's postulates and the "one-gene/one-enzyme/one-function" concept, has successfully identified numerous causal relationships, such as smoking with lung cancer and asbestos with mesothelioma [21] [22] [23]. However, the growing recognition that factors at multiple biological levels—from genes and proteins to behavioral patterns and social determinants—influence health and disease has challenged this dominant epidemiological paradigm [21]. Complex chronic diseases such as diabetes, cancer, and Alzheimer's disease rarely follow simple linear causality but instead emerge from intricate networks of interacting elements characterized by dynamic feedback loops, reciprocal relations, and non-linear interactions [22] [23] [24]. This article objectively compares these competing philosophies—linear causality versus complex network interactions—examining their foundational principles, methodological approaches, and applications in drug development and precision medicine.
The limitations of reductionist approaches become evident when considering diseases like obesity, where causative factors span endogenous elements (genes, epigenetic factors), individual-level behaviors (diet, exercise), neighborhood-level influences (food availability, walking environment), and even national-level policies (agricultural support, food programs) [21]. Similarly, Alzheimer's disease manifests with highly variable presentation influenced by genetic inheritance, age at onset, sex differences, environmental exposures, and polygenic risk scores, making simple linear models inadequate for capturing its complexity [24]. This recognition has catalyzed a methodological shift toward complex systems dynamic computational models that can better represent the multiscale, interactive nature of disease pathogenesis [21] [22].
Conceptual Foundations: Core Principles and Philosophical Frameworks
Linear Causality Model
The linear causality model, rooted in 19th-century germ theory and Koch's postulates, operates on the fundamental principle that specific, isolatable agents cause corresponding diseases [22]. This reductionist approach seeks to isolate independent factors that directly cause disease states, using conceptual frameworks such as the sufficient-component causal model and counterfactual paradigm to establish causation [21]. The methodology predominantly employs regression-based models—including multivariable and multilevel regression—that assess relationships between "independent" variables and disease outcomes while controlling for potential confounders [21] [22]. This paradigm conceptualizes diseases as having singular, actionable causes and forms the philosophical foundation for much of contemporary evidence-based medicine, particularly in establishing causal relationships between risk factors and diseases [21].
Complex Network Interaction Model
The complex network interaction model conceptualizes diseases as emergent properties of perturbed biological systems rather than isolated malfunctions [23] [25]. This framework recognizes that cellular networks operate through specific laws and principles, and that phenotypes result from perturbations to these interconnected systems [23]. The approach utilizes interactome networks—simplified representations of cellular systems as nodes (biological components) and edges (interactions between them)—to model disease pathogenesis [23] [26]. Methodologically, it employs computational approaches such as agent-based modeling, network diffusion algorithms, and machine learning applied to multiscale data [21] [27] [26]. This philosophy fundamentally challenges linear causality by acknowledging reciprocal relationships (where causes and effects influence each other), dynamic feedback loops, and the absence of predictable parametric relations in biological systems [21].
Table 1: Fundamental Principles of Each Approach
| Principle | Linear Causality Model | Complex Network Interaction Model |
|---|---|---|
| Causal Structure | Unidirectional, deterministic | Multidirectional, probabilistic |
| System View | Reductionist, focusing on isolated components | Holistic, focusing on system interactions |
| Disease Emergence | Direct consequence of specific causes | Emergent property of perturbed networks |
| Temporal Dynamics | Static relationships | Dynamic, feedback-driven evolution |
| Intervention Strategy | Target specific causal factors | Modulate network properties |
Visualizing the Conceptual Differences
The following diagram illustrates the fundamental structural differences between linear and network-based disease models:
Methodological Comparison: Analytical Approaches and Techniques
Data Requirements and Experimental Design
Linear approaches primarily rely on controlled experimental designs that isolate variables of interest, with data structures optimized for regression analyses [21]. These methods typically require clearly defined independent and dependent variables, with careful attention to confounding factors [21]. In contrast, network medicine integrates diverse omics datasets—genomics, transcriptomics, proteomics, metabolomics—to construct comprehensive interactome networks that capture the complexity of biological systems [23] [28]. The multiscale interactome approach further incorporates biological functions into protein-protein interaction networks, creating hierarchical networks that span from molecular interactions to organism-level phenotypes [26]. The integration of imaging data with omics datasets represents another advancement, enabling researchers to link brain-level functional and structural changes to molecular-level alterations in neurodegenerative diseases like Alzheimer's [24].
Key Analytical Techniques
Linear methodologies employ regression-based techniques including multivariable regression, logistic regression, and multilevel (hierarchical) models that estimate the effects of specific variables while controlling for others [21]. While these methods are powerful for identifying isolated relationships, they struggle with reciprocal relations between exposures and outcomes, discontinuous relations, and changes in relationships over time [21]. Network-based approaches utilize diverse computational methods including agent-based modeling (simulating individual agents and their interactions) [21], network diffusion profiles (using random walks to model effect propagation) [26], and machine learning algorithms (such as Random Forest and XGBoost) that incorporate network topology and protein features to predict biomarker potential [27].
Table 2: Methodological Approaches and Applications
| Methodology | Primary Techniques | Key Applications | Limitations |
|---|---|---|---|
| Regression-Based Models | Multivariable regression, multilevel modeling | Isolating independent risk factors, controlling for confounders | Poor handling of reciprocal relationships, non-linear dynamics |
| Agent-Based Modeling | Computer simulation of individual agents with defined interaction rules | Modeling population-level emergence from individual interactions, obesity epidemiology | Computational intensity, parameter specification challenges |
| Network Diffusion | Biased random walks on multiscale networks | Predicting drug-disease treatments, identifying therapeutic mechanisms | Network completeness, edge weight optimization |
| Machine Learning Integration | Random Forest, XGBoost on network features | Predictive biomarker identification, cancer signaling analysis | Interpretability challenges, training data requirements |
Experimental Workflow for Network-Based Drug Discovery
The following diagram outlines a generalized experimental workflow for identifying drug treatments using network-based approaches:
Performance Comparison: Quantitative Findings and Experimental Evidence
Predictive Accuracy in Drug-Disease Treatment
A systematic evaluation of the multiscale interactome approach demonstrated significant improvements in predicting drug-disease treatments compared to molecular-scale interactome methods that only consider physical interactions between proteins [26]. The multiscale approach achieved an AUROC of 0.705 versus 0.620 (+13.7%) and average precision of 0.091 versus 0.065 (+40.0%) [26]. This enhanced performance was particularly notable for entire drug classes such as hormones, which rely heavily on biological functions and cannot be accurately represented by approaches considering only physical interactions [26]. The study analyzed nearly 6,000 approved treatments spanning almost every category of human anatomy, exceeding the largest prior network-based study by tenfold [26].
Biomarker Discovery and Validation
Network-based approaches have demonstrated particular utility in identifying predictive biomarkers for targeted cancer therapies. The MarkerPredict framework, which integrates network motifs and protein disorder information, classified 3,670 target-neighbor pairs with 32 different machine learning models achieving 0.7-0.96 leave-one-out-cross-validation accuracy [27]. By defining a Biomarker Probability Score (BPS) as a normalized summative rank of the models, the method identified 2,084 potential predictive biomarkers for targeted cancer therapeutics, with 426 classified as biomarkers by all four calculations [27]. This systematic approach demonstrates how network properties can enhance biomarker discovery beyond linear association studies.
Quantitative Comparison of Methodological Performance
Table 3: Experimental Performance Metrics Across Methodologies
| Performance Metric | Linear Regression Models | Multiscale Network Approach | Improvement |
|---|---|---|---|
| Drug-Disease Prediction AUROC | 0.620 | 0.705 | +13.7% |
| Drug-Disease Prediction Average Precision | 0.065 | 0.091 | +40.0% |
| Recall@50 | 0.264 | 0.347 | +31.4% |
| Biomarker Prediction Accuracy (LOOCV) | N/A | 0.7-0.96 | N/A |
| Therapeutic Coverage | Limited to direct targets | Extensive, including functional matches | Substantial |
The Scientist's Toolkit: Essential Research Reagents and Solutions
Implementing network approaches requires specialized computational resources and datasets. The following table outlines essential research reagents and their applications in complex disease modeling:
Table 4: Essential Research Reagents for Network Medicine
| Resource Category | Specific Examples | Function/Application |
|---|---|---|
| Protein Interaction Databases | SIGNOR, ReactomeFI, Human Cancer Signaling Network | Provide physical and functional interaction data for network construction |
| Biological Function Annotations | Gene Ontology (GO) terms | Annotate biological processes, molecular functions, and cellular components |
| Biomarker Databases | CIViCmine, DisProt | Provide validated biomarker information for model training and validation |
| ORFeome Collections | Human ORFeome libraries | Enable high-throughput interactome mapping using standardized open reading frames |
| Machine Learning Frameworks | Random Forest, XGBoost | Implement classification of potential biomarkers based on network features |
| Network Analysis Tools | FANMOD, Cytoscape | Identify network motifs and visualize complex biological networks |
Experimental Protocols for Key Methodologies
Multiscale Interactome Construction and Analysis
The multiscale interactome methodology integrates physical interactions between 17,660 human proteins (387,626 edges) with 9,798 biological functions from Gene Ontology (34,777 edges between proteins and biological functions, 22,545 edges between biological functions) [26]. The protocol involves: (1) compiling drug-target interactions (8,568 edges connecting 1,661 drugs to human proteins) and disease-protein associations (25,212 edges connecting 840 diseases to disrupted human proteins); (2) constructing the multiscale network by connecting proteins to biological functions according to established hierarchies; (3) computing diffusion profiles using biased random walks with optimized edge weights (wdrug, wdisease, wprotein, wbiological function, whigher-level biological function, wlower-level biological function); (4) comparing drug and disease diffusion profiles to predict treatments and identify relevant proteins and biological functions [26].
Predictive Biomarker Identification Using Network Motifs
The MarkerPredict protocol for identifying predictive biomarkers in oncology includes: (1) extracting three-nodal network motifs (triangles) from cancer signaling networks using FANMOD; (2) annotating intrinsically disordered proteins (IDPs) using DisProt, AlphaFold (pLLDT<50), and IUPred (score>0.5); (3) creating training sets from literature-curated positive controls (established predictive biomarkers) and negative controls (proteins not in biomarker databases); (4) training Random Forest and XGBoost machine learning models on network topological features and protein disorder annotations; (5) calculating Biomarker Probability Scores (BPS) as normalized summative ranks across models; (6) validating predictions through literature mining and experimental follow-up [27].
Discussion: Clinical Implications and Future Directions
Translation to Precision Medicine
The transition from linear causality to network-based approaches has profound implications for precision medicine. Network medicine provides a systems-level framework for understanding how genetic variants interact with environmental factors to produce disease phenotypes [28] [24]. In Alzheimer's disease research, integrating imaging data with omics datasets has enabled the identification of disease subtypes and the development of more personalized risk assessments [24]. Similarly, in oncology, network-based biomarker discovery approaches like MarkerPredict offer the potential to identify patients who will respond to targeted therapies, sparing others from unnecessary side effects [27]. The multiscale interactome's ability to explain treatment mechanisms even when drugs seem unrelated to the diseases they treat represents a significant advance in pharmacological understanding [26].
Limitations and Methodological Challenges
Despite their promise, network-based approaches face several important limitations. Incomplete interactome maps remain a fundamental challenge, as current networks likely miss important interactions and context-specificities [23] [28]. The sheer complexity of biological systems presents interpretability challenges, particularly when integrating across multiple biological scales [28]. Additionally, network medicine requires sophisticated computational infrastructure and specialized expertise that may not be readily available in all research settings [25] [28]. For linear models, their relative simplicity, established statistical frameworks, and interpretability maintain their utility for many research questions, particularly when investigating specific, well-defined causal pathways [21].
Emerging Innovations and Future Perspectives
The field of network medicine is rapidly evolving with several promising directions. The incorporation of temporal dynamics through longitudinal network analysis could capture disease progression more accurately than static networks [25] [28]. Advanced machine learning methods, particularly deep learning architectures, are being integrated with network approaches to enhance predictive power [27] [28]. Innovative modeling frameworks, including quantum mechanics-based approaches that represent individual health states as quantum superposition states, offer novel ways to capture the uncertainty and heterogeneity inherent in disease processes [29]. The continued development of more comprehensive and context-specific interactome maps will further enhance the resolution and accuracy of network-based disease models [23] [28].
The comparison between linear causality and complex network interactions in disease modeling reveals a nuanced landscape where each approach offers distinct advantages and limitations. Linear models provide conceptual clarity and statistical rigor for investigating specific causal pathways, while network approaches better capture the systemic complexity of multifactorial diseases. Rather than a wholesale replacement of one paradigm by the other, the future of disease research likely lies in their strategic integration—using linear approaches for well-defined causal questions and network methods for understanding system-level dynamics. This complementary use of methodologies, leveraging the respective strengths of each, promises to accelerate progress toward more effective, personalized approaches for understanding, preventing, and treating complex diseases.
The classical reductionist approach in biological research has historically focused on the identification and characterization of isolated components of living organisms. While successful in cataloging individual biological elements, this perspective has proven inadequate for clarifying the complex interaction mechanisms between components and predicting how alterations in single or multiple elements affect entire system dynamics [30]. In contrast, systems biology represents a fundamental shift in perspective, aiming to understand biology at the system level through functional analysis of the structure and dynamics of cells and organisms [30]. This discipline focuses not on isolated components, but on the complex network of interactions between genes, proteins, metabolites, and other biomolecules that collectively give rise to biological function [30].
The emergence of systems biology as a practical discipline has been catalyzed by the data revolution brought about by high-throughput omics technologies. These technologies enable comprehensive, large-scale analysis of diverse biomolecular layers, including the genome, epigenome, transcriptome, and proteome [31]. The ability to simultaneously examine entire systems rather than single genes or proteins has transformed our approach to understanding health and disease, particularly for complex disorders known to be caused by combinations of genetic, environmental, immunological, and neurological factors [30]. This article examines how these technological advances have enabled a systems-level understanding of biology, comparing the performance of different approaches and methodologies that form the foundation of modern biological research.
The Enabling Technologies: A Multi-Layered View of Biology
High-throughput omics technologies have revolutionized biological research by providing unprecedented insights into the complexity of living systems at multiple molecular levels [32]. The integration of data from these complementary technologies provides a more holistic and representative understanding of the complex molecular mechanisms that underpin biology [31].
Table 1: High-Throughput Omics Technologies and Their Applications
| Omics Type | Key Technologies | Biological Focus | Research Applications |
|---|---|---|---|
| Genomics | Next-generation sequencing (NGS) | DNA structure, function, and variation | Identifying genetic mutations, understanding disease genetics [32] [31] |
| Epigenomics | DNA methylation analysis, ChIP-Seq | Modifications of DNA and DNA-associated proteins | Studying gene regulation, understanding epigenetic influences on disease [32] [31] |
| Transcriptomics | RNA sequencing (RNA-Seq) | RNA transcripts and gene expression regulation | Analyzing gene expression changes, understanding regulatory mechanisms [32] [31] |
| Proteomics | Mass spectrometry, affinity-based methods | Protein identification, quantification, and modification | Understanding protein functions, identifying biomarkers and therapeutic targets [32] [31] |
| Metabolomics | NMR spectroscopy, mass spectrometry | Metabolite profiles and metabolic pathways | Identifying metabolic changes, understanding pathways and disease mechanisms [32] |
| Single-cell Omics | Single-cell sequencing | Cellular heterogeneity at multiple molecular levels | Investigating cellular heterogeneity, understanding cell functions in development and disease [32] |
The true power of these technologies emerges through their integration in a multi-omics approach. Studying each molecular layer in isolation can only reveal part of the biological picture, while bringing all these different layers together provides a more complete understanding of human biology and disease [31]. For example, combining genomics and proteomics allows researchers to directly link genotype to phenotype, while integrating transcriptomics and proteomics provides insights into how gene expression affects protein function and phenotypic outcomes [31]. This integrative approach is essential for unraveling the complexity of cellular processes and disease mechanisms [32].
Comparative Analysis: Systems Biology Versus Reductionist Biomarker Approaches
Traditional reductionist approaches and modern systems biology methods differ fundamentally in their philosophy, methodology, and applications. The reductionist perspective has typically addressed the study of living organisms by focusing on isolated components rather than the complex system as a whole [30]. In contrast, systems biology employs a holistic perspective that examines the simultaneous interactions of multiple system elements [30].
Philosophical and Methodological Differences
The reductionist approach to biomarker discovery and therapeutic development typically focuses on single molecules or linear signaling pathways when identifying diagnostic biomarkers or drug targets [30] [33]. This "single-target-based" drug development approach has proven notably less effective for complex diseases, with lower probability of success and higher risk in addressing underlying disease biology [34]. The fundamental limitation of this approach lies in its inability to capture the emergent properties of biological systems that arise from complex networks of interactions [34].
Systems biology, conversely, recognizes that biological function is rarely regulated by a single molecule, but rather emerges from complex interactions among a cell's distinct components [30]. This perspective employs network analysis as a primary tool for representing biological relationships, leveraging mathematical tools from Graph Theory to understand system behavior [30]. In this framework, groups of interacting molecules that regulate discrete functions form biomodules whose interrelations create complex networks [30].
Performance Comparison in Disease Research
The practical differences between these approaches become evident when examining their application to complex disease research. A systems biology study of colorectal cancer (CRC) exemplifies the power of the network-based approach. Researchers identified 848 differentially expressed genes between normal and cancerous tissue, then constructed a protein-protein interaction (PPI) network which revealed 99 hub genes with high connectivity [33]. Clustering analysis dissected this network into seven interactive modules, providing a systems-level view of the molecular interactions driving CRC progression [33]. This approach identified several genes with high centrality in the PPI network that contribute to CRC progression, including CCNA2, CD44, and ACAN, which were found to correlate with poor patient prognosis [33].
Similarly, a systems biology approach to COVID-19 research demonstrated the advantages of network-based analysis over single-target methods. By collecting 757 genes associated with COVID-19 from literature databases and constructing a PPI network, researchers identified hub proteins with high connectivity [35]. Subsequent controllability analysis of directed COVID-19 signaling pathways revealed driver genes with high control power over the network state [35]. Expression data analysis confirmed that these hub and driver genes showed significant differential expression between COVID-19 and control groups, and perhaps more importantly, exhibited different expression correlation patterns between the two groups [35]. This network-based approach enabled the identification of potential drug combinations that could target multiple nodes in the disease network simultaneously [35].
Table 2: Comparison of Reductionist vs. Systems Biology Approaches in Disease Research
| Aspect | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Analytical Focus | Single molecules or linear pathways [30] | Complex networks and interactions [30] |
| Therapeutic Strategy | "Single-target" drugs [34] | Multi-targeted therapies and drug combinations [34] [35] |
| Network Perspective | Limited consideration of interactions | Centrality and controllability analysis [33] [35] |
| Biomarker Discovery | Individual molecular biomarkers | Network biomarkers and correlation patterns [35] |
| Handling of Complexity | Often inadequate for complex diseases | Specifically designed for complex, multifactorial diseases [30] [34] |
| Clinical Success Rate | Lower for complex diseases [34] | Potential to increase probability of success in clinical trials [34] |
Experimental Protocols and Data Integration Methodologies
The implementation of systems biology approaches relies on sophisticated experimental protocols and computational methodologies designed to handle the complexity and volume of multi-omics data. This section details key experimental workflows and the critical challenge of data integration in multi-omics studies.
Network Analysis and Hub Gene Identification
A representative protocol for network-based analysis involves several standardized steps, as demonstrated in the colorectal cancer and COVID-19 studies [33] [35]:
Data Acquisition: Retrieval of gene expression data from public repositories such as the Gene Expression Omnibus (GEO). For the CRC study, this involved obtaining datasets containing both normal and colorectal cancer tissue samples [33].
Differential Expression Analysis: Identification of significantly differentially expressed genes (DEGs) using statistical packages in R/Bioconductor. In the CRC study, this analysis revealed 848 DEGs [33].
Network Construction: Building protein-protein interaction (PPI) networks using databases such as STRING, which integrates known and predicted protein interactions [33] [35]. The COVID-19 study began with 757 literature-derived genes associated with the disease [35].
Centrality Analysis: Using network analysis software such as Cytoscape and Gephi to identify hub genes based on network centrality measures [33]. The CRC study identified 99 hub genes through this approach [33].
Module Detection: Applying clustering algorithms (e.g., k-means) to identify interactive modules or communities within the larger network [33]. The CRC network was dissected into seven interactive modules [33].
Functional Enrichment: Conducting gene-set enrichment analysis based on Gene Ontology (GO) and KEGG pathway databases to identify biological functions and pathways associated with gene groups [33].
Survival Analysis: Examining the prognostic value of identified hub genes using survival analysis tools such as GEPIA [33].
This workflow enables the transition from individual gene analysis to a systems-level understanding of disease mechanisms, identifying key nodes in biological networks that may serve as effective therapeutic targets [33] [35].
Network analysis workflow in systems biology: This diagram illustrates the sequential process from data acquisition to biomarker identification, highlighting key computational tools and databases used at each stage.
Data Integration Challenges and Solutions
Data integration represents one of the most significant challenges in multi-omics research, as it involves combining different omics datasets with varying characteristics, scales, and levels of noise [32] [31]. The optimal integration strategy depends on several factors, including the biological question being addressed, the type and quality of the data, and the experimental context [31].
Two fundamental computational approaches have emerged for multi-omics integration:
Similarity-based methods focus on identifying common patterns, correlations, and shared pathways across different omics datasets. These include:
- Correlation analysis to evaluate relationships between different omics levels
- Clustering algorithms (e.g., hierarchical clustering, k-means) to group similar data points
- Network-based approaches such as Similarity Network Fusion (SNF) to construct integrated networks [32]
Difference-based methods emphasize detecting unique features and variations between omics levels, including:
- Differential expression analysis to identify significant changes between states
- Variance decomposition to partition variance into omics-specific components
- Feature selection methods (e.g., LASSO, Random Forests) to select relevant features from each omics dataset [32]
Popular integration algorithms include Multi-Omics Factor Analysis (MOFA), which uses Bayesian factor analysis to identify latent factors responsible for variation across multiple omics datasets, and Canonical Correlation Analysis (CCA), which identifies linear relationships between two or more omics datasets [32].
Addressing Batch Effects and Data Quality
A critical technical challenge in large-scale omics studies is the presence of batch effects - technical biases introduced when combining datasets from different sources or experiments [36]. These effects can hinder quantitative comparison of independently acquired datasets and potentially confound biological conclusions.
Recent methodological advances have addressed this challenge through sophisticated batch-effect correction methods. The Batch-Effect Reduction Trees (BERT) algorithm represents a significant innovation in this area, designed specifically for handling incomplete omic profiles [36]. BERT employs a tree-based data integration framework that decomposes data integration tasks into a binary tree of batch-effect correction steps, using established methods like ComBat and limma at each node [36].
Table 3: Performance Comparison of Data Integration Methods
| Method | Handling of Missing Data | Computational Efficiency | Ability to Handle Covariates | Key Advantages |
|---|---|---|---|---|
| BERT | Retains up to 5 orders of magnitude more numeric values than HarmonizR [36] | Up to 11× runtime improvement over alternatives [36] | Considers covariates and reference measurements [36] | Hierarchical approach, handles severely imbalanced conditions [36] |
| HarmonizR | Unique removal (UR) approach introduces data loss [36] | Lower efficiency compared to BERT [36] | Limited handling of design imbalance [36] | Imputation-free framework, employs matrix dissection [36] |
| MOFA | Handles missing values through probabilistic modeling | Moderate computational demands | Integrates multiple omics with sample covariates | Unsupervised approach, identifies latent factors [32] |
| CCA | Requires complete cases or imputation | Computationally efficient for large datasets | Limited covariate integration | Identifies correlated features across omics layers [32] |
In benchmark evaluations on simulated and experimental data with up to 5000 datasets, BERT demonstrated superior performance in retaining numeric values (minimizing data loss) while improving computational efficiency [36]. This approach is particularly valuable for large-scale integrative studies where data completeness and quality are major concerns.
Visualization Approaches for Multi-Omic Data
Effective visualization is essential for interpreting the complex datasets generated in systems biology research. Traditional heatmaps and color-coded representations have been widely used for pairwise comparisons of omics datasets, but these approaches have limitations when comparing three or more conditions [37].
Three-Way Comparison Methodology
A novel color-coding approach based on the HSB (hue, saturation, brightness) color model has been developed to facilitate intuitive visualization of three-way comparisons [37]. This method employs the circular nature of the hue component to map possible distributions of three compared values onto color space:
Hue Assignment: The three compared values are assigned specific hue values from the circular hue range (e.g., red, green, and blue) [37].
Color Calculation: The resulting hue representing the three-way comparison is calculated according to the distribution of the three compared values:
- If all three values are identical, the resulting color is white
- If two values are identical and one is different, the resulting hue corresponds to the characteristic hue of the differing value
- If all three values are different, the resulting hue is selected from a color gradient between the two most distant values according to the relative position of the third value [37]
Saturation Encoding: The saturation of the color reflects the amplitude of the numerical difference between the two most distant values according to a scale of interest [37].
Brightness Modulation: The brightness can be set to maximum by default or used to encode additional information about the three-way comparison [37].
This visualization approach was applied to three-way comparisons of metabolite profiles from capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS) analysis of mouse liver samples, successfully highlighting different types of value distributions across experimental conditions [37].
Three-way comparison visualization method: This diagram outlines the process for visualizing three-way comparisons of omics data using the HSB color model, highlighting different distribution patterns and their corresponding color representations.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Implementing systems biology approaches requires a diverse set of research reagents, computational tools, and platforms. The table below details essential resources used in the featured studies and the broader field.
Table 4: Essential Research Reagents and Platforms for Systems Biology
| Resource Type | Specific Tool/Platform | Function and Application |
|---|---|---|
| Data Resources | Gene Expression Omnibus (GEO) | Public repository of functional genomics data [33] |
| Interaction Databases | STRING Database | Resource of known and predicted protein-protein interactions [33] [35] |
| Pathway Databases | KEGG Pathways | Collection of pathway maps representing molecular interactions and networks [35] |
| Network Analysis Software | Cytoscape | Open-source platform for complex network visualization and analysis [30] [33] |
| Statistical Analysis | R/Bioconductor | Programming environment for statistical analysis of omics data [33] |
| Batch Effect Correction | BERT (Batch-Effect Reduction Trees) | High-performance method for data integration of incomplete omic profiles [36] |
| Multi-Omics Integration | OmicsNet, NetworkAnalyst | Platforms for visual analysis of biological networks integrating multiple omics types [32] |
| Sequencing Platforms | Next-generation sequencing (NGS) | High-throughput DNA and RNA sequencing for genomic and transcriptomic analysis [32] |
| Proteomics Platforms | Mass spectrometry | Identification and quantification of proteins and their modifications [32] |
| Metabolomics Platforms | CE-TOFMS, NMR spectroscopy | Comprehensive analysis of metabolite profiles [37] [32] |
The data revolution driven by high-throughput omics technologies has fundamentally transformed biological research, enabling a comprehensive systems-level understanding of living organisms. The shift from reductionist approaches to network-based systems biology represents more than just a methodological change - it constitutes a fundamental rethinking of how we study health and disease [30] [34]. By focusing on the complex interactions between biological components rather than isolated elements, systems biology provides a more accurate and productive framework for understanding biological complexity [30].
The integration of multi-omics data through advanced computational methods has created unprecedented opportunities for biomarker discovery and therapeutic development [32]. This is particularly valuable for complex diseases like cancer, COVID-19, and autoimmune disorders, where understanding the interplay between genetic mutations, gene expression changes, protein modifications, and metabolic shifts is critical for developing effective treatments [33] [32] [35]. The continued evolution of single-cell multi-omics technologies and spatial omics approaches promises to further enhance our resolution of biological systems, revealing cellular heterogeneity and tissue organization in unprecedented detail [38] [31].
As systems biology continues to mature, its integration with emerging technologies like artificial intelligence and machine learning will likely accelerate the discovery process, enabling more predictive models of human disease and more effective therapeutic interventions [32] [34]. However, researchers must remain mindful of challenges such as data shift, under-specification, overfitting, and the "black box" nature of some complex models [31]. Despite these challenges, the systems biology paradigm, powered by high-throughput omics technologies, is positioned to remain a key pillar of biological research and drug development, ultimately advancing more effective, personalized therapeutic strategies [34].
Operationalizing Systems Biology: Tools, Workflows, and Real-World Applications in Biomarker Research
The field of biomedical research is defined by a fundamental methodological divide. On one side lies the long-established reductionist approach, which focuses on isolating and studying individual biomarkers—single molecules, such as a specific protein or gene, that indicate a biological state or disease condition. While powerful for developing targeted diagnostic tests, this approach is inherently limited in its capacity to represent the complex, interconnected nature of living systems. In contrast, systems biology embraces a holistic philosophy, seeking to understand biological phenomena through the lens of complex, interacting networks. It integrates diverse data types—multi-omics, clinical, and environmental—to construct computational models that can simulate system-wide behavior, predict emergent properties, and ultimately guide more effective therapeutic interventions [39] [40]. This guide provides a objective comparison of the key tools and databases that enable the systems biology approach, framing them against the backdrop of traditional biomarker methods.
The limitations of a purely reductionist framework are evident in areas like ovarian cancer research. While biomarkers like CA-125 and HE4 are valuable, their diagnostic performance is often suboptimal due to low specificity; CA-125 levels, for instance, can elevate in many non-cancerous conditions [41]. Machine learning models that integrate multiple biomarkers have demonstrated superior performance, achieving AUC values exceeding 0.90, yet they still operate primarily on correlative associations rather than mechanistic understanding [41]. Systems biology toolkits aim to move beyond correlation to causation by building predictive, mechanistic models of human physiology, such as digital twins of drug pharmacokinetics and pharmacodynamics in diseases like type 2 diabetes [39].
Table 1: Core Conceptual Comparison: Systems Biology vs. Reductionist Biomarker Approaches
| Feature | Systems Biology Approach | Reductionist Biomarker Approach |
|---|---|---|
| Core Philosophy | Holistic, network-oriented | Targeted, single-variable oriented |
| Primary Focus | Emergent properties of interacting components | Individual molecules or pathways |
| Typical Data | Multi-omics (genomics, proteomics, etc.), clinical, environmental | Focused biomarker measurements (e.g., serum protein levels) |
| Key Methodology | Computational modeling, network analysis, simulation | Statistical association, hypothesis testing on single biomarkers |
| Model Output | Predictive, mechanistic simulations (e.g., digital twins) | Diagnostic or prognostic scores (e.g., ROMA index in ovarian cancer) [41] |
| Strengths | Captures complexity, enables prediction and simulation, provides mechanistic insight | Clinically established, often simpler to implement and interpret |
| Limitations | Computationally intensive, requires diverse data, complex model validation | May miss critical system-level interactions and feedback loops |
Comparative Analysis of Key Pathway Databases
Pathway databases are foundational to systems biology, providing the structured knowledge of biological interactions upon which networks and models are built. The choice of database is not merely a technicality; it directly influences the results of statistical enrichment analysis and predictive modeling, a factor often overlooked in reductionist analyses [42].
A systematic benchmarking study demonstrated that equivalent pathways from different databases yield disparate results in enrichment analysis. Furthermore, the performance of machine learning models for patient classification and survival analysis showed a significant, dataset-dependent impact based on the pathway resource used [42]. This variability underscores the importance of database selection. To mitigate this, integrative resources like MPath have been developed. MPath merges analogous pathways from KEGG, Reactome, and WikiPathways, creating a unified resource that in some cases improves prediction performance and yields more biologically consistent enrichment results [42].
Table 2: Quantitative and Qualitative Comparison of Major Pathway Databases
| Database | Pathway Count | Reaction Count | Compound Count | Key Features & Scope | Key Advantages | Key Disadvantages |
|---|---|---|---|---|---|---|
| KEGG | 179 modules, 237 maps [43] | 8,692 [43] | 16,586 [43] | Broad coverage, includes modules and maps; strong in metabolism and xenobiotics degradation [43] | Well-known, widely used; includes non-metabolic pathways | Licensing can be restrictive; pathway conceptualization can be overly broad [44] |
| MetaCyc | 1,846 base pathways, 296 super pathways [43] | 10,262 [43] | 11,991 [43] | Non-redundant, experimentally elucidated pathways; strong in plant, fungal, and bacterial metabolism [43] | High-quality curation; includes taxonomic range; fewer unbalanced reactions | Smaller compound database than KEGG |
| Reactome | 2,119 pathways [42] | Not explicitly listed | Not explicitly listed | Detailed, hierarchical pathway knowledge; strong in human biology and signal transduction [42] | Sophisticated visualization; extensive cross-links to other databases [44] | Can be highly detailed, which may not always be necessary |
| WikiPathways | 409 pathways [42] | Not explicitly listed | Not explicitly listed | Community-curated, open-access platform for biological pathway models [42] | Fully open and community-driven; rapidly updated | Smaller overall size compared to Reactome and KEGG |
| MPath (Integrative) | 2,896 total pathways (including 129 analogs, 26 super pathways) [42] | Not explicitly listed | Not explicitly listed | A merged resource combining KEGG, Reactome, and WikiPathways, unifying equivalent pathways [42] | Reduces database-specific bias; can improve prediction performance and result consistency [42] | Merging pathways from different sources is a complex process |
Benchmarking Toolkit Performance: Software and Modeling Paradigms
The computational engine of systems biology is its software ecosystem, which enables the creation, simulation, and analysis of biological network models. These tools can be broadly categorized into those used for dynamical modeling (often using ordinary differential equations) and those for constraint-based modeling (such as Flux Balance Analysis).
A key innovation in this space is the move towards programmatic modeling, which combines computational modeling with software engineering best practices. Using general-purpose programming languages like Python, researchers can encode models as executable code, which enhances modularity, testing, documentation, and reproducibility [40]. This paradigm shift, supported by tools like COBRApy for constraint-based analysis and Tellurium for dynamical modeling, facilitates collaborative model development and more robust, shareable research outcomes [39] [40].
Table 3: Comparison of Key Software Tools for Systems Biology Modeling
| Software Tool | Primary Modeling Type | Core Function | Language/Environment | Key Features |
|---|---|---|---|---|
| COBRA Toolbox / COBRApy | Constraint-Based | Quantitative prediction of cellular metabolism [45] | MATLAB / Python [45] | Flux balance analysis, flux variability analysis; genome-scale metabolic modeling [39] |
| Tellurium | Dynamical | Reproducible dynamical modeling of biological networks [39] | Python [39] | Integrated environment for simulating biochemical networks; supports standard formats like SBML and SED-ML [39] |
| libRoadRunner | Dynamical | High-performance simulation of SBML models [39] | C/C++ with Python interface [39] | Uses LLVM for ultra-fast simulation; benchmark for performance in computational biology [39] |
| sbmlutils | Both (Utility) | Python utilities for working with SBML models [39] | Python [39] | Simplifies model creation, manipulation, annotation, and provides file converters [39] |
| PK-DB | Pharmacokinetic (PK) | FAIR-compliant open database for pharmacokinetics data [39] | Database / Python | Enables reproducible PBPK/PD modeling and individualized simulations [39] |
Experimental Protocols for Tool and Database Benchmarking
To objectively assess the performance of different pathway databases and modeling tools, researchers employ standardized benchmarking protocols. The following methodologies, derived from the literature, provide a framework for comparative analysis.
Protocol 1: Benchmarking Pathway Database Impact on Enrichment Analysis [42]
- Data Retrieval: Obtain pathway data from major databases (e.g., KEGG, Reactome, WikiPathways) and convert them into a consistent format, such as the Gene Matrix Transposed (GMT) file format.
- Dataset Selection: Select multiple -omics datasets from public repositories like The Cancer Genome Atlas (TCGA) to ensure results are not dataset-specific.
- Statistical Enrichment: Perform enrichment analysis on each dataset using each pathway resource. Common methods include the hypergeometric test, Gene Set Enrichment Analysis (GSEA), and Signaling Pathway Impact Analysis (SPIA).
- Result Comparison: Systematically compare the lists of significantly enriched pathways generated by each database. Metrics for comparison can include the number of significant pathways, the degree of overlap between lists, and the biological plausibility of the results.
- Integration Test: Create an integrative database (e.g., MPath) by merging analogous pathways from the primary resources and repeat the enrichment analysis to determine if integration yields more consistent or biologically informative results.
Protocol 2: Evaluating Predictive Modeling Performance with Pathway Data [42]
- Pathway Activity Scoring: Use a method like single-sample GSEA (ssGSEA) to calculate pathway activity scores for each sample in a clinical cohort.
- Model Training: Train machine learning models (e.g., for patient survival prediction or disease classification) using the pathway activity scores derived from different pathway databases as features.
- Performance Assessment: Evaluate and compare the performance of the models using appropriate metrics, such as AUC (Area Under the Curve) for classification or C-index for survival analysis. Statistical tests should be used to determine if performance differences between databases are significant.
Protocol 3: Building and Simulating a Programmatic Model [40]
- Model Definition: Use a Python package like
sbmlutilsortelluriumto define a computational model programmatically. This involves specifying model components (species, parameters), reactions, and initial conditions directly in code. - Simulation and Analysis: Execute the model to run simulations, perform parameter scans, or conduct sensitivity analysis using the programmatic environment's native functions.
- Export and Validation: Export the model to a standardized format like the Systems Biology Markup Language (SBML) to validate the model in other software tools, ensuring interoperability and reproducibility [39] [46].
- Reproducibility Packaging: Package the entire modeling and analysis workflow, including code, data, and environment specifications, to enable others to reproduce the results exactly.
Essential Research Reagent Solutions
The following table details key resources, both computational and data-oriented, that constitute the essential "research reagent solutions" for a modern systems biology toolkit.
Table 4: Key Research Reagent Solutions for Systems Biology
| Item Name | Type | Function in Research |
|---|---|---|
| Pathway Databases (KEGG, Reactome, etc.) | Knowledgebase | Provide curated, computable representations of biological pathways for network analysis and model building [43] [42]. |
| SBML (Systems Biology Markup Language) | Model Format | Serves as a lingua franca for representing computational models of biological processes, ensuring exchangeability between different software tools [47] [46]. |
| COBRApy | Software Library | Enables constraint-based reconstruction and analysis of metabolic networks at the genome scale, including prediction of metabolic fluxes [39] [45]. |
| Digital Twin Platform (e.g., PBPK/PD models) | Computational Model | Creates patient-specific physiological models to predict individual responses to drugs and diseases, enabling personalized treatment strategies [39]. |
| PK-DB | Data Resource | A FAIR-compliant database for pharmacokinetics data, supporting the parameterization and validation of pharmacokinetic models [39]. |
| Programmatic Modeling Environment (e.g., Python) | Software Framework | Provides a flexible, code-based environment for building, simulating, and analyzing models, enhancing reproducibility and collaboration [40]. |
Visualizing Workflows and Signaling Pathways
The following diagrams, generated with Graphviz DOT language, illustrate core workflows and concepts in systems biology analysis.
Systems Biology Analysis Workflow
Pathway Integration Creates MPath
Notch Signaling Pathway
The systems biology toolkit, comprising multi-omics integration, sophisticated network modeling, and AI/ML, represents a paradigm shift from traditional reductionist biomarker approaches. The comparative data presented in this guide demonstrates that the choice of specific resources—from pathway databases to software platforms—has a measurable impact on analytical outcomes. While reductionist methods provide clarity and focus on individual components, systems biology offers the powerful ability to model complex interactions and predict emergent behaviors. The future of biomedical research and drug development lies in the strategic combination of both approaches, leveraging the precision of biomarkers within the predictive, systems-level framework provided by computational models and digital twins.
Bottom-Up vs. Top-Down vs. Middle-Out Analytical Approaches
In the evolving landscape of biological research, the debate between holistic systems biology and traditional reductionist approaches is central to advancing our understanding of complex diseases. Reductionist methods have long focused on isolating individual biomarkers, but this can overlook the complex network interactions that define living systems. Systems biology, employing top-down, bottom-up, and middle-out analytical approaches, seeks to understand these systems as a whole. This guide provides an objective comparison of these three foundational frameworks, underpinned by experimental data and their specific applications in modern research and drug development.
Core Analytical Approaches at a Glance
The table below summarizes the defining characteristics, objectives, and primary applications of the three main analytical approaches in systems biology.
| Approach | Core Principle | Primary Objective | Ideal Application Context | Data Flow Direction |
|---|---|---|---|---|
| Top-Down | Hypothesis-driven; starts with high-level, system-wide data to identify key modules or players. [48] [49] | Uncover emergent properties and identify critical, high-value targets from a holistic starting point. [48] | Analyzing complex 'omics' data (e.g., from transcriptomics, proteomics) to find signatures of disease. [48] [49] | From system-level phenomena down to specific molecular components. [49] |
| Bottom-Up | Data-driven; starts by assembling detailed components into a system-wide model. [48] [49] | Construct a comprehensive, mechanistic model of a system from its fundamental parts. [48] [49] | Building detailed, predictive models for in-silico testing of perturbations (e.g., drug effects). [49] | From molecular components up to an integrated system model. [49] |
| Middle-Out | A hybrid, rational strategy that starts from a key functional subsystem. [48] [50] | Engineer systems with improved performance by balancing theoretical design and empirical evolution. [50] | Projects requiring system improvement or upgrading existing systems where a full top-down restart is not feasible. [51] [52] [50] | From a critical middle layer, expanding both upward to system goals and downward to components. [52] |
Detailed Experimental Protocols and Workflows
Top-Down Proteomics Analysis
The top-down approach in proteomics involves analyzing intact proteins to gain a comprehensive view of proteoforms, including those with post-translational modifications (PTMs). [53]
Experimental Protocol:
- Sample Preparation: Purify proteins from a cell lysate or tissue sample using techniques like liquid chromatography (LC) to reduce complexity.
- Intact Protein Analysis: Introduce the intact protein ions into a high-resolution mass spectrometer (e.g., FT-ICR or Orbitrap) via electrospray ionization (ESI). [53]
- Gas-Phase Fragmentation: Isolate a specific protein ion and subject it to gas-phase fragmentation. Electron-Capture Dissociation (ECD) or Electron-Transfer Dissociation (ETD) are preferred as they randomly cleave the peptide backbone and preserve labile PTMs. [53]
- Data Analysis: The high-mass-accuracy fragment ions (c and z ions) are used to deduce the complete protein sequence and pinpoint the location of any modifications. [53]
Bottom-Up Proteomics Analysis
The bottom-up strategy digests proteins into peptides prior to mass spectrometry analysis, making it the most mature and widely used method for high-throughput protein identification. [53]
Experimental Protocol:
- Protein Digestion: Digest the purified protein or complex protein mixture with a protease (e.g., trypsin) to generate a peptide mixture. [53]
- Peptide Separation: Separate the complex peptide mixture using multi-dimensional liquid chromatography (e.g., ion-exchange coupled to reversed-phase LC). [53]
- Tandem MS (MS/MS) Analysis: As peptides elute, they are ionized and analyzed. A specific peptide ion is isolated and fragmented using Collision-Induced Dissociation (CID), which preferentially cleaves peptide bonds. [53]
- Database Search: The resulting fragmentation pattern (b and y ions) is matched against theoretical spectra in a protein database to identify the original protein. [53]
Middle-Out Systems Engineering
In systems engineering, the middle-out approach is applied when upgrading or improving an existing system, using operational scenarios to drive both higher-level requirements and lower-level component design. [52]
Methodology:
- Start with Operational Analysis: Begin the analysis in the middle of the system hierarchy by defining key operational scenarios that the improved system must perform. [52]
- Define Requirements: Use the operational analysis to create and validate high-level system requirements. [52]
- Component Decomposition: Decompose the system requirements downward to identify and specify the physical components and their interfaces needed to support the operational scenarios. [52]
Comparative Performance Data
The following table summarizes quantitative and qualitative data comparing the three approaches across key performance metrics, particularly in the context of proteomics and model-building.
| Performance Metric | Top-Down Proteomics | Bottom-Up Proteomics | Middle-Out Engineering |
|---|---|---|---|
| Sequence Coverage | High - Provides complete protein sequence and full PTM characterization. [53] | Limited - Identifies only a fraction of the total peptide population. [53] | Focused - Based on the scope of the selected mid-level subsystem. [52] |
| PTM Analysis | Excellent - ECD/ETD preserves labile PTMs, allowing precise localization. [53] | Poor - Labile PTMs are often lost during CID fragmentation. [53] | Context-Dependent - Inherits characteristics based on the chosen approach for the subsystem. |
| Throughput & Maturity | Lower throughput; less mature technology and data analysis tools. [53] | High throughput; mature, widely used, and automated. [53] | Moderate - More efficient than a full bottom-up restart but requires careful planning. [51] [52] |
| Handling Complex Mixtures | Challenging for highly complex samples due to current technology limits. [53] | Excellent - The benchmark for analyzing complex protein digests (e.g., cell lysates). [53] | Effective - Designed to handle complexity by constraining the problem space. [52] [50] |
| Primary Instrumentation | High-resolution MS (FT-ICR, Orbitrap) with ECD/ETD. [53] | Ion traps, Q-TOF, TOF-TOF with CID. [53] | Model-based systems engineering tools (e.g., CORE). [52] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of these approaches relies on a suite of specialized reagents and computational tools.
| Item Name | Function / Application | Relevant Approach |
|---|---|---|
| High-Resolution Mass Spectrometer (e.g., FT-ICR, Orbitrap) | Enables accurate mass measurement of intact proteins and their fragments for top-down sequencing. [53] | Top-Down |
| Electron-Transfer Dissociation (ETD) Reagents | Chemical reagents that facilitate ETD fragmentation, preserving post-translational modifications. [53] | Top-Down |
| Trypsin (Protease) | Enzymatically cleaves proteins into peptides for bottom-up mass spectrometry analysis. [53] | Bottom-Up |
| Multi-Dimensional Liquid Chromatography (LC) System | Separates complex peptide mixtures to reduce sample complexity and increase protein identification in bottom-up proteomics. [53] | Bottom-Up |
| COBRA (Constraint-Based Reconstruction and Analysis) Toolbox | A computational toolbox for building, simulating, and analyzing genome-scale metabolic models in bottom-up systems biology. [49] | Bottom-Up |
| STRATA Methodology / CORE Tool | A model-based systems engineering methodology and tool for managing requirements, behavior, and physical architecture in complex projects. [52] | Middle-Out |
| Stable Isotope Labels | Used for quantitative proteomics and metabolic flux analysis in both top-down and bottom-up frameworks. [53] [49] | Top-Down & Bottom-Up |
The choice between top-down, bottom-up, and middle-out is not about finding a single "best" method, but rather about selecting the right tool for the research question and context. [51] [54]
- Use a Top-Down approach when the goal is to identify critical, high-value targets from a holistic starting point, such as discovering biomarker signatures from multi-omics data. [48] [49]
- Employ a Bottom-Up approach when the objective is to build a detailed, predictive model of a known system for in-silico testing and hypothesis validation. [48] [49]
- Adopt a Middle-Out strategy for complex engineering or optimization tasks where a full top-down restart is impractical, and a focused, rational hybrid approach can yield superior, integrated results. [52] [50]
The future of biological research and drug development lies in the intelligent integration of these approaches, leveraging their complementary strengths to bridge the gap between reductionist biomarker discovery and a truly systemic understanding of disease.
Mesenchymal stromal/stem cells (MSCs) have emerged as a promising therapeutic tool for various conditions, from autoimmune diseases to tissue repair, with over 13,300 registered clinical trials as of 2023 [55]. Despite encouraging preclinical results and a favorable safety profile, the clinical translation of MSC therapies has been hampered by inconsistent efficacy and variable outcomes [56] [57]. This inconsistency primarily stems from the inherent heterogeneity of MSC populations, which manifests at multiple levels: differences in tissue sources (bone marrow, adipose tissue, umbilical cord), donor-specific variations (age, health status), manufacturing processes, and intercellular functional diversity [55] [56] [58].
The traditional reductionist approach to drug discovery, which focuses on modulating single molecular targets identified through in vitro assays, has proven inadequate for addressing the complex heterogeneity of living cell therapies [59]. This case study examines how integrated Systems Biology (SysBio) and Artificial Intelligence (AI), collectively termed SysBioAI, are overcoming these limitations by providing a holistic, data-driven framework for understanding and controlling MSC heterogeneity, thereby enabling more consistent and effective stem cell therapies [60].
The heterogeneity of MSC-based Advanced Therapy Medicinal Products (ATMPs) originates from multiple sources, which can be broadly categorized as shown in Table 1 [56] [58].
Table 1: Primary Sources of Heterogeneity in MSC-Based Therapies
| Category | Specific Factors | Impact on MSC Product |
|---|---|---|
| Donor Attributes | Age, sex, genetics, health status, body mass index [55] [56] | Influences MSC phenotype, proliferation capacity, differentiation potential, and secretory profile [55] [58] |
| Tissue Source | Bone marrow, adipose tissue, umbilical cord, dental pulp, placental tissue [55] [56] | Distinct gene expression profiles, differentiation biases, and immunomodulatory properties [56] [57] |
| Manufacturing & Preparation | Isolation methods, culture media composition, serum supplements, oxygen tension, passaging number, cryopreservation protocols [56] [57] | Affects cell viability, potency, senescence, immunogenicity, and clinical functionality [58] [57] |
This multidimensional variability makes it extremely challenging to predict clinical performance using conventional quality control measures that rely on a limited set of surface markers (CD105, CD73, CD90) and differentiation assays [55] [56]. The reductionist paradigm fails to capture the complex, interconnected networks that determine MSC functionality in the dynamic in vivo environment [59] [60].
SysBioAI: An Integrated Analytical Framework
SysBioAI represents a paradigm shift from reductionism to a holistic, integrative approach. Systems Biology employs computational and mathematical modeling to understand complex biological systems as integrated wholes, analyzing interactions between genes, proteins, and cellular pathways [60]. When combined with Artificial Intelligence—particularly machine learning (ML) and deep learning (DL) algorithms—this framework gains the ability to identify complex, non-linear patterns within large-scale, multi-dimensional datasets [60] [61].
The synergy of SysBioAI is particularly powerful for addressing MSC heterogeneity because it can [60]:
- Integrate diverse multi-omics data (genomics, transcriptomics, proteomics, metabolomics)
- Model the dynamic, non-linear relationships between molecular determinants and clinical outcomes
- Predict cellular behavior and therapeutic potency with unprecedented accuracy
- Identify critical biomarker signatures that correlate with clinical efficacy
Figure 1: SysBioAI Integrative Analytical Framework. The model shows how multi-omics data and clinical parameters are processed through combined machine learning and systems biology approaches to generate predictive models and biomarkers.
Comparative Analysis: Reductionist vs. SysBioAI Approaches
The fundamental differences between traditional reductionist methods and the emerging SysBioAI paradigm are substantial, with distinct implications for addressing MSC heterogeneity, as detailed in Table 2.
Table 2: Systematic Comparison of Reductionist versus SysBioAI Approaches
| Analytical Characteristic | Reductionist Approach | SysBioAI Approach |
|---|---|---|
| Primary Focus | Single genes, proteins, or pathways [59] | Complex, interconnected networks and systems [60] |
| Data Integration Capacity | Limited, typically analyzes one data type at a time [59] | High, integrates multi-omics data simultaneously [60] [61] |
| Heterogeneity Handling | Poor, seeks to minimize or ignore variability [59] | Robust, explicitly models and accounts for variability [60] |
| Predictive Power for Clinical Outcomes | Low, frequently fails to predict in vivo efficacy [59] [57] | High, identifies complex patterns correlating with outcomes [60] |
| Mechanism of Action (MoA) Elucidation | Limited to linear, simplified pathways [59] | Comprehensive, reveals non-linear, dynamic interactions [60] |
| Experimental Design | Hypothesis-driven, targeted assays [59] | Discovery-driven, untargeted multi-omics [60] [61] |
| Therapeutic Optimization Strategy | One-dimensional (e.g., optimize single protein activity) [59] | Multi-dimensional (e.g., optimize complex functional signatures) [60] |
The limitations of the reductionist approach are evident in the history of drug discovery, where programs beginning with compound selection based on single-protein biochemical assays have largely failed for complex diseases [59]. This is particularly problematic for MSC therapies, where functional properties emerge from complex, dynamic interactions within the cells and with their microenvironment [60].
Experimental Protocols for SysBioAI Analysis
Implementing SysBioAI analysis for MSC characterization involves a multi-stage workflow that generates and integrates diverse data types. The following protocols outline key experimental and computational methodologies.
Multi-Omics Data Generation Protocol
Objective: To generate comprehensive molecular profiling data from heterogeneous MSC populations for subsequent SysBioAI analysis [60] [61].
Methodology:
- Sample Preparation: Obtain MSCs from different tissue sources (e.g., bone marrow, adipose tissue, umbilical cord) and donors. Culture under standardized conditions and at various passages to capture process-related heterogeneity [56] [58].
- Genomic Sequencing: Perform whole-genome sequencing to identify genetic variants and epigenetic modifications (DNA methylation) that contribute to functional heterogeneity [61].
- Single-Cell RNA Sequencing (scRNA-seq): Apply scRNA-seq to profile transcriptomic heterogeneity within and between MSC populations, identifying distinct functional subpopulations [56] [60].
- Proteomic Analysis: Utilize mass spectrometry-based proteomics to quantify protein expression and post-translational modifications across different MSC batches [61].
- Metabolomic Profiling: Employ LC-MS/MS to characterize metabolic pathways and small molecule signatures associated with MSC potency [61].
Quality Control: Implement strict batch effect correction, normalization procedures, and replicate sampling to ensure data quality and reproducibility [60] [61].
Computational Integration and Modeling Protocol
Objective: To integrate multi-omics data streams and build predictive models of MSC therapeutic potency [60] [61].
Methodology:
- Data Preprocessing: Normalize heterogeneous data types using appropriate transformations and handle missing values through imputation algorithms [61].
- Feature Selection: Apply unsupervised learning algorithms (e.g., autoencoders) for dimensionality reduction and identification of most informative features [61].
- Network Analysis: Construct biological networks using systems biology tools to map interactions between genes, proteins, and metabolites across different MSC populations [60].
- Predictive Modeling: Train supervised machine learning algorithms (e.g., Random Forest, Support Vector Machines) using integrated multi-omics features as inputs and clinical efficacy measures as outputs [60] [61].
- Model Validation: Employ cross-validation and independent test sets to assess model performance and prevent overfitting [61].
Output: Validated predictive models that identify molecular signatures correlating with specific MSC functional properties and clinical outcomes [60].
Figure 2: SysBioAI Experimental Workflow. The diagram outlines the key stages from multi-omics data generation through computational analysis to practical application for quality control and prediction.
Key Research Reagent Solutions for SysBioAI Implementation
The successful implementation of SysBioAI analysis requires specialized reagents and computational tools. Table 3 details essential solutions for researchers in this field.
Table 3: Essential Research Reagent Solutions for SysBioAI in Stem Cell Research
| Reagent/Tool Category | Specific Examples | Function in SysBioAI Analysis |
|---|---|---|
| Single-Cell RNA Sequencing Kits | 10X Genomics Chromium, SMART-seq reagents [56] | Enable transcriptomic profiling of heterogeneous MSC populations at single-cell resolution [56] [60] |
| Mass Spectrometry Reagents | TMT/Label-free proteomics kits, metabolomics extraction kits [61] | Facilitate comprehensive proteomic and metabolomic characterization of MSC functional states [61] |
| Cell Culture Media Systems | Defined, xeno-free MSC media with consistent composition [56] [57] | Reduce batch-to-batch variability introduced by culture conditions during expansion [56] |
| Flow Cytometry Panels | Extended surface marker panels beyond standard ISCT criteria [56] [58] | Enable high-dimensional immunophenotyping correlated with functional properties [58] |
| Bioinformatics Platforms | Seurat, Scanpy, CellPhoneDB, XGBoost, TensorFlow [60] [61] | Provide computational infrastructure for data integration, network analysis, and machine learning [60] [61] |
| Public Data Repositories | TCGA, GEO, ArrayExpress, Human Cell Atlas [61] | Offer reference datasets for model training and validation across diverse cell populations [61] |
The integration of Systems Biology and Artificial Intelligence represents a transformative approach to overcoming the critical challenge of heterogeneity in MSC-based therapies. By moving beyond the limitations of reductionist biomarker strategies, SysBioAI enables a holistic understanding of the complex molecular networks that determine therapeutic efficacy [60]. This paradigm shift allows researchers to model MSC heterogeneity as a measurable variable rather than an uncontrollable nuisance, paving the way for predictive potency assays and consistently effective stem cell products [60] [61].
As SysBioAI methodologies continue to evolve, they promise to accelerate the development of personalized stem cell therapies tailored to individual patient profiles and specific disease contexts [60] [61]. This patient-centric, data-driven framework establishes a new paradigm for precision and regenerative medicine, potentially unlocking the full clinical potential of mesenchymal stem cells that has remained elusive under traditional analytical approaches [60].
The study of complex neurological disorders has undergone a paradigm shift, moving from traditional reductionist models to integrative systems-level approaches. Reductionist methods, which focus on isolating single biomarkers or linear pathways, often fail to capture the multifaceted etiology of conditions like autism spectrum disorder (ASD) and attention-deficit/hyperactivity disorder (ADHD) [62] [30]. In contrast, systems biology leverages computational tools and network analysis to map the complex, interacting web of genetic, molecular, and clinical factors that underlie these disorders [30] [33]. This case study objectively compares these two methodological frameworks, demonstrating how network analysis not only identifies robust, multi-node biomarkers but also reveals distinct neurobiological subtypes that are invisible to conventional diagnostic criteria [62]. We provide supporting experimental data and detailed protocols to guide researchers in deploying these powerful analytical techniques.
Systems Biology vs. Reductionist Approaches: A Conceptual and Practical Comparison
The core distinction between these frameworks lies in their scope and underlying philosophy. The following table summarizes their key differences.
Table 1: Comparative Analysis of Research Frameworks in Neuroscience
| Aspect | Reductionist (Biomarker-Focused) Approach | Systems Biology (Network Analysis) Approach |
|---|---|---|
| Core Philosophy | Studies components in isolation to identify single, causative factors [30]. | Studies systems as a whole, focusing on interactions and emergent properties [30]. |
| View of Disease | Linear causality from a primary molecular defect. | A network perturbation arising from dynamic interactions across multiple levels [30] [33]. |
| Primary Methodology | Targeted assays (e.g., ELISA, PCR) for specific molecules. | High-throughput 'omics' integration (genomics, proteomics) and computational modeling [30] [33]. |
| Data Output | Quantification of a limited set of predefined biomarkers. | System-wide maps of interactions (e.g., Protein-Protein Interaction networks) [30] [33]. |
| Biomarker Identification | Aims for a single, specific diagnostic or prognostic marker. | Identifies hub genes and interactive modules central to the network structure [33]. |
| Strength | Simplicity, well-established protocols, and straightforward interpretation. | Holistic view, ability to discover novel, unexpected relationships, and subtyping [62] [30]. |
| Limitation | Incomplete picture, inability to model complex interactions or identify subtypes [30]. | Computational complexity, requires large datasets, and sophisticated bioinformatics expertise [30]. |
Recent research underscores the power of the systems approach. For instance, analysis of over 123,000 structural MRI scans identified two distinct neurobiological subtypes of ADHD—delayed brain growth (DBG-ADHD) and prenatal brain growth (PBG-ADHD)—which exhibit significant disparities in functional organization at the network level despite being indistinguishable by conventional criteria [62].
Experimental Protocol for Network Analysis in Neurological Disorders
The following workflow provides a detailed methodology for applying systems biology to deconvolute complex neurological disorders, synthesizing protocols from key studies [62] [33].
Data Acquisition and Preprocessing
- Data Collection: Retrieve relevant high-dimensional data from public repositories like the Gene Expression Omnibus (GEO) for transcriptomics or the UK Biobank for neuroimaging data. For a gene expression study, this would involve downloading datasets from patients and healthy controls [33].
- Data Cleaning and Normalization: This critical step involves handling missing values, identifying and treating outliers, and normalizing the data to remove technical artifacts. Common tasks include log-transformation of gene expression data and spatial normalization of brain images [63].
Differential Analysis and Network Construction
- Identification of Differentially Expressed Genes (DEGs): Using statistical packages in R/Bioconductor, perform an analysis (e.g., LIMMA for microarray data) to identify genes with significant expression changes between case and control groups. A study on colorectal cancer, for example, identified 848 DEGs using this method [33].
- Network Reconstruction: Construct a Protein-Protein Interaction (PPI) network using databases such as STRING. The list of DEGs is input into the database to extract known and predicted interactions, which form the edges of the network [33].
Network Centrality and Cluster Analysis
- Centrality Analysis: Import the PPI network into visualization software like Cytoscape or Gephi. Calculate network centrality metrics (e.g., degree, betweenness) to identify hub genes—highly connected nodes that are potentially functionally critical. One study identified 99 hub genes via this analysis [33].
- Module Detection: Use clustering algorithms (e.g., k-means, MCODE) within Cytoscape to partition the network into densely connected sub-networks or modules. These modules often correspond to distinct biological functions or pathways [33].
Functional Enrichment and Survival Analysis
- Gene Ontology (GO) and Pathway Analysis: Perform functional enrichment analysis using GO and KEGG pathway databases on the hub genes and individual modules. This determines the biological processes, molecular functions, and pathways that are significantly over-represented in the disease network [33].
- Survival Analysis: To validate the clinical relevance of identified hub genes, use a tool like GEPIA to perform survival analysis. This tests whether high or low expression of a hub gene is correlated with significant differences in patient survival rates [33].
The following diagram illustrates this integrated experimental workflow:
Key Experimental Data and Findings
The systems biology approach yields multi-faceted, quantitative data that can be summarized for clear comparison.
Table 2: Key Findings from Network Analysis in Neurological and Neuropsychiatric Disorders
| Disorder | Key Finding | Data Type | Experimental Support |
|---|---|---|---|
| ADHD [62] | Identification of two neurobiological subtypes (DBG-ADHD, PBG-ADHD) with distinct network-level functional organization. | Neuroimaging (MRI) | Analysis of over 123,000 structural MRI scans using standardized brain charts. |
| ASD & ADHD [62] | Personalized Brain Network (PBN) profiles reliably predict individual cognitive, behavioral, and sensory phenomena. | Connectome-based Prediction Modeling | Use of connectome-based prediction modeling and normative modeling on large-scale datasets (e.g., UK Biobank, N=8,086). |
| Colorectal Cancer [33] | Identification of 99 hub genes from a PPI network; survival analysis confirmed 3 hub genes (CCNA2, CD44, ACAN) linked to poor prognosis. | Transcriptomics (Gene Expression) | Differential expression analysis, PPI network centrality, and survival analysis (GEPIA). |
Table 3: The Scientist's Toolkit: Essential Research Reagents and Solutions
| Item / Resource | Function / Application | Specific Examples / Notes |
|---|---|---|
| R/Bioconductor [33] | Open-source software for statistical computing and analysis of genomic data. | Used for differential gene expression analysis [33]. |
| STRING Database [33] | A database of known and predicted protein-protein interactions. | Used to reconstruct the initial PPI network from a list of genes [33]. |
| Cytoscape / Gephi [30] [33] | Open-source software platforms for complex network visualization and analysis. | Used for network visualization, calculation of centrality metrics, and module detection [33]. |
| Gene Ontology (GO) & KEGG [33] | Databases for functional annotation and pathway enrichment analysis. | Used to determine the biological significance of hub genes and network modules [33]. |
| GEPIA [33] | An online tool for survival analysis based on gene expression data from cancer patients. | Used to validate the prognostic value of identified hub genes [33]. |
| fMRI/DTI [62] | Neuroimaging techniques to measure brain activity and structural connectivity. | Used to build personalized brain network architectures and identify "neural fingerprints" [62]. |
Visualization of a Hypothesized Disease Network
To illustrate the output of such an analysis, the following diagram depicts a simplified, hypothesized network for a complex neurological disorder. Hub genes, representing potential therapeutic targets, are highlighted.
This case study demonstrates a clear and objective comparison between reductionist and systems biology approaches. The data and protocols detailed herein confirm that network analysis provides a superior framework for deconvoluting complex neurological disorders. By moving beyond single biomarkers to model the entire interactive system, researchers can achieve a more holistic understanding of disease mechanisms, identify robust multi-gene signatures, and discover previously hidden patient subtypes. This paradigm is foundational to the emerging field of precision neurodiversity, which seeks to develop tailored interventions based on an individual's unique brain network architecture, ultimately celebrating neurological variation as a source of human strength [62]. For the drug development professional, this translates into more precise target identification and stratified clinical trials, increasing the likelihood of therapeutic success.
The field of drug discovery is undergoing a fundamental transformation, shifting from traditional reductionist approaches toward integrative systems biology frameworks. Reductionist methods have historically focused on single biomarkers—such as individual genes or proteins—to guide therapeutic development, providing valuable but often fragmented insights into complex disease mechanisms [64]. In contrast, modern systems biology approaches leverage multi-omics data, artificial intelligence, and network-based analyses to capture the intricate interactions within biological systems [65]. This paradigm shift is particularly crucial for addressing complex diseases like cancer, neurodegenerative disorders, and chronic conditions, where disease pathogenesis emerges from dynamic interactions across multiple biological scales rather than isolated molecular defects.
The limitations of single-marker approaches have become increasingly apparent in precision oncology. While biomarkers like EGFR mutations in non-small cell lung cancer and HER2 amplification in breast cancer have revolutionized targeted therapies, tumor heterogeneity and adaptive resistance mechanisms often undermine their long-term efficacy [66]. This recognition has catalyzed the development of dual-biomarker strategies that simultaneously target oncogenic drivers while modulating the immune microenvironment, representing a more holistic approach to therapeutic intervention [67]. This article provides a comprehensive comparison of reductionist versus systems biology approaches in biomarker discovery, examining their respective applications in identifying driver genes and developing effective combination therapies for complex diseases.
Comparative Analysis of Biomarker Discovery Approaches
Table 1: Fundamental characteristics of reductionist versus systems biology approaches
| Characteristic | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Analytical Focus | Single biomarkers (genes, proteins) | Multi-omics networks and pathways |
| Therapeutic Strategy | Monotherapies targeting individual drivers | Combination therapies addressing multiple mechanisms |
| Data Integration | Limited contextual integration | Multi-modal data fusion (genomics, proteomics, network topology) |
| Experimental Validation | Targeted assays with high specificity | High-throughput screening with computational prioritization |
| Clinical Translation | Straightforward but limited applicability | Complex but potentially higher clinical impact |
| Representative Methods | PCR, immunohistochemistry, single-gene sequencing | Machine learning on signaling networks, multi-omics integration, AI-powered simulations |
Reductionist approaches have demonstrated significant clinical utility in contexts where disease pathogenesis is driven by clearly identifiable molecular alterations. For example, EGFR inhibitors in EGFR-mutant lung cancer and BRAF inhibitors in BRAF-mutant melanoma exemplify the success of targeted therapeutic strategies [66]. These approaches benefit from straightforward diagnostic methodologies, relatively clear regulatory pathways, and well-defined mechanisms of action. However, their limitations become apparent when addressing complex, multifactorial diseases where tumor heterogeneity and adaptive resistance mechanisms frequently lead to treatment failure [67].
Systems biology frameworks address these limitations by incorporating network-based analyses that capture the complex interactions within biological systems. The MarkerPredict platform exemplifies this approach by integrating network motifs and protein disorder properties to identify predictive biomarkers with machine learning models achieving 0.7-0.96 LOOCV (Leave-One-Out Cross-Validation) accuracy [27]. This method identified 2,084 potential predictive biomarkers for targeted cancer therapeutics by analyzing three signaling networks, demonstrating the power of systems-level approaches to generate comprehensive biomarker panels that would remain undetected through reductionist methods [27]. Similarly, multi-omics integration enables researchers to develop comprehensive molecular maps of diseases by combining genomics, transcriptomics, proteomics, and metabolomics data, thereby identifying complex marker combinations that traditional methods might overlook [65] [8].
Quantitative Comparison of Approach Performance
Table 2: Performance metrics of reductionist versus systems biology approaches in precision oncology
| Performance Metric | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Biomarker Discovery Rate | Limited by hypothesis-driven design | 32 different ML models identifying 426 high-confidence biomarkers [27] |
| Predictive Accuracy | Variable context-dependent performance | 0.7-0.96 LOOCV accuracy range [27] |
| Clinical Benefit Rate | 20-40% in biomarker-matched populations | 53% disease control rate in dual-matched therapy [67] |
| Therapeutic Durability | Often limited by resistance mechanisms | Exceptional responders with PFS >23 months observed [67] |
| Model Interpretability | High mechanistic clarity | Variable; requires specialized analytical frameworks |
| Patient Coverage | Limited to specific molecular subgroups | Potential for broader application across heterogeneous populations |
The quantitative comparison reveals distinct advantages and limitations for each approach. Reductionist strategies demonstrate consistent performance in specific clinical contexts where disease biology is well-characterized and driven by dominant molecular alterations. For example, EGFR mutation testing in NSCLC successfully identifies patients who benefit from EGFR inhibitors, with response rates exceeding 60% in biomarker-matched populations [66].
Systems biology approaches demonstrate superior performance in addressing complex disease mechanisms and identifying combination therapy opportunities. A clinical study of dual-matched therapy—where both gene-targeted agents and immune checkpoint inhibitors were selected based on distinct biomarkers—achieved a 53% disease control rate despite 29% of patients having undergone ≥3 prior therapies [67]. Notably, three patients (~18%) achieved prolonged progression-free survival (23.4+, 33.0, and 59.7 months) and overall survival (23.4+, 43.6, and 62.1+ months), demonstrating the potential for exceptional outcomes when therapies are matched to comprehensive biomarker profiles [67].
The integration of artificial intelligence further enhances systems biology approaches by enabling the identification of complex, non-linear relationships within high-dimensional biomedical data. Machine learning algorithms, particularly Random Forest and XGBoost models, have demonstrated robust performance in biomarker discovery, with the MarkerPredict framework achieving high accuracy through the analysis of network-based properties and protein structural features [27]. These computational approaches can process diverse data streams, including genetic markers, protein profiles, and medical imaging, to generate comprehensive predictive insights that extend beyond basic diagnosis to anticipate treatment responses and outcomes [8].
Experimental Protocols for Biomarker Discovery and Validation
Reductionist Approach: Linear Validation of Single Biomarkers
The reductionist approach to biomarker validation follows a sequential, hypothesis-driven pathway with clearly defined stages:
- Candidate Identification: Literature review and preliminary data analysis to select potential biomarker candidates based on known disease mechanisms.
- Assay Development: Establishment of targeted detection methods such as PCR, immunohistochemistry, or ELISA with optimization for sensitivity and specificity.
- Analytical Validation: Determination of assay precision, accuracy, sensitivity, specificity, and reproducibility using reference standards and control samples.
- Clinical Validation: Evaluation of biomarker-disease association in well-defined patient cohorts, typically through retrospective studies.
- Regulatory Approval: Submission of validated data to regulatory agencies for diagnostic approval.
- Clinical Implementation: Integration of the biomarker into routine clinical practice for patient stratification or treatment selection.
This linear workflow benefits from standardized methodologies and clear regulatory pathways but is constrained by its reliance on pre-existing knowledge of disease mechanisms, potentially overlooking novel biomarkers operating outside established pathways.
Systems Biology Approach: Integrated Multi-Omics Workflow
Systems biology employs an integrated, cyclical workflow that combines high-throughput data generation with computational analysis:
Systems Biology Multi-Omics Workflow
The protocol initiates with multi-omics data collection from diverse molecular layers, including genomics, transcriptomics, proteomics, and metabolomics [65]. These data are then integrated through computational pipelines that construct molecular networks and identify dysregulated pathways. The MarkerPredict implementation exemplifies this stage by analyzing three signaling networks (CSN, SIGNOR, ReactomeFI) and incorporating protein disorder predictions from DisProt, AlphaFold, and IUPred databases [27].
Machine learning models are subsequently trained on these integrated datasets to identify complex patterns associated with therapeutic response. The MarkerPredict framework employed both Random Forest and XGBoost algorithms, utilizing topological information from signaling networks and protein annotations to optimize model decision-making [27]. Model outputs are then synthesized into composite scores, such as the Biomarker Probability Score (BPS), to prioritize candidates for experimental validation [27].
The final stage involves experimental validation of prioritized biomarkers using in vitro and in vivo models, with results informing subsequent iterations of the discovery cycle. This iterative refinement process enables continuous improvement of biomarker panels and enhances their predictive performance.
Signaling Pathways in Biomarker-Driven Therapeutics
Reductionist Model: Linear Signaling Pathway
Linear Pathway for Single-Target Therapy
Reductionist approaches conceptualize signaling as linear pathways with defined inputs and outputs, enabling straightforward therapeutic targeting but failing to capture the complexity and adaptability of biological systems. This model underpins many successful targeted therapies but ultimately proves inadequate for addressing the robust nature of cellular networks that rapidly develop resistance through pathway reactivation or bypass mechanisms.
Systems Biology Model: Network-Based Signaling
Network-Based Signaling for Combination Therapy
Systems biology represents signaling as interconnected networks with redundant pathways and regulatory loops that maintain homeostasis despite therapeutic perturbation. This framework reveals critical network properties, such as the enrichment of intrinsically disordered proteins (IDPs) in network triangles, which function as information processing hubs and represent promising biomarker candidates [27]. The recognition of these network features enables the rational design of combination therapies that simultaneously target multiple nodes, preventing resistance development through network adaptation.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential research reagents and platforms for biomarker discovery and validation
| Category | Specific Tools/Platforms | Research Applications |
|---|---|---|
| Multi-Omics Technologies | Single-cell RNA sequencing, Spatial transcriptomics, High-throughput proteomics, Metabolomics platforms | Comprehensive molecular profiling across biological scales [65] [66] |
| Computational Tools | Random Forest/XGBoost classifiers, Network analysis software (Cytoscape), IUPred/AlphaFold for disorder prediction | Machine learning-based biomarker classification and network modeling [27] |
| Signaling Network Databases | CSN (Cancer Signaling Network), SIGNOR, ReactomeFI, Human Cancer Signaling Network | Contextualizing biomarkers within biological pathways [27] |
| Validation Assays | Liquid biopsy platforms (ctDNA, CTCs), Multiplexed immunofluorescence, Imaging mass cytometry, Organoid/co-culture systems | Experimental validation of computational predictions [67] [66] |
| AI-Powered Platforms | Digital twin simulations, Virtual patient platforms, QSP (Quantitative Systems Pharmacology) models | Clinical trial optimization and biomarker validation [68] |
The modern biomarker researcher requires access to diverse technological platforms that span molecular profiling, computational analysis, and experimental validation. Multi-omics technologies form the foundation of systems biology approaches, enabling the generation of comprehensive molecular datasets that capture disease heterogeneity across biological scales [65] [66]. These profiling technologies are complemented by computational tools that extract meaningful patterns from high-dimensional data, with machine learning classifiers like Random Forest and XGBoost demonstrating particular utility in biomarker prioritization [27].
The integration of biomarker discovery with biological context depends on signature network databases that catalog molecular interactions and pathway relationships. The MarkerPredict framework utilized three signaling networks with distinct topological characteristics to contextualize potential biomarkers within their functional networks [27]. Finally, advanced validation assays including liquid biopsy platforms and complex model systems enable the translation of computational predictions into biologically meaningful insights with clinical applicability [67] [66].
The comparison between reductionist and systems biology approaches reveals a compelling trajectory for future biomarker discovery. While reductionist methods provide specificity and regulatory tractability for well-characterized molecular targets, systems biology approaches offer comprehensive coverage of complex disease mechanisms and enhanced potential for addressing therapeutic resistance. The clinical success of dual-matched therapies—achieving a 53% disease control rate in heavily pretreated patients—demonstrates the significant potential of systems-guided approaches [67].
The emerging paradigm in biomarker discovery integrates elements from both frameworks, leveraging the precision of reductionist validation while incorporating the contextual understanding provided by systems biology. This integrated approach utilizes AI-powered platforms to navigate the complexity of multi-omics data while maintaining focus on clinically actionable biomarkers [8] [68]. As these technologies mature, they promise to accelerate the development of effective combination therapies that address the multifaceted nature of complex diseases, ultimately advancing the goal of precision medicine across diverse patient populations.
Navigating Implementation Challenges: Data, Modeling, and Translational Hurdles in Systems Biomarker Discovery
Addressing Data Heterogeneity, Standardization, and Reproducibility Issues
The pursuit of reliable biomarkers for complex diseases represents a critical frontier in modern medicine, where two competing research philosophies collide: reductionism versus systems biology. The reductionist approach, dominating early biomarker discovery, isolates and studies individual molecular components in controlled environments. While this method has yielded significant discoveries, it often fails to capture the complex, interconnected reality of biological systems, leading to biomarkers that perform poorly in real-world clinical applications. In contrast, systems biology embraces biological complexity by studying biomarkers as components within vast, interacting networks, mirroring the true nature of cellular signaling and regulation [27] [24].
This paradigm shift occurs against a challenging backdrop of pervasive data heterogeneity and reproducibility crises in biomedical research. Biomarker data originates from diverse sources—genomic sequencing, proteomic assays, clinical records, and medical imaging—each with distinct formats, scales, and technical artifacts [69] [70]. Without rigorous standardization, these datasets become incompatible, preventing meaningful integration and validation. Simultaneously, the reproducibility of research findings remains a significant concern, particularly as artificial intelligence and machine learning become more prevalent in biomarker discovery [71] [70]. These challenges necessitate a fundamental re-evaluation of how biomarker research is conducted, from experimental design to data sharing.
Comparative Analysis: Reductionist vs. Systems Biology Approaches
The table below summarizes the fundamental differences between reductionist and systems biology approaches to biomarker research, highlighting their distinct strategies for addressing data challenges.
Table 1: Comparison of Reductionist and Systems Biology Approaches to Biomarker Research
| Aspect | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Philosophical Basis | Studies components in isolation | Studies systems as integrated networks |
| Data Handling | Focuses on single data types; minimal integration challenges | Integrates multi-omics data (genomics, proteomics, etc.); requires robust data harmonization [72] [73] |
| Network Considerations | Lacks network context; views biomarkers as independent entities | Incorporates network topology and motif analysis (e.g., triangle motifs in signaling networks) [27] |
| Reproducibility Challenges | Technically simpler to reproduce but may lack clinical relevance | Complex workflows require detailed documentation and standardization for reproducibility [71] |
| Clinical Translation | Often fails due to oversimplification of biology | Higher potential by capturing complex disease mechanisms [24] |
| Technology Requirements | Standard molecular biology tools | Requires advanced computational infrastructure, AI/ML, and multi-omics platforms [72] [73] |
The Impact on Biomarker Validation and Clinical Translation
The choice between these approaches significantly impacts biomarker validation. Reductionist methods often produce biomarkers that demonstrate excellent analytical performance in controlled settings but fail to predict therapeutic responses in heterogeneous patient populations. This occurs because they overlook compensatory pathways and network adaptations that emerge in intact biological systems [27] [24].
Systems biology frameworks address these limitations by incorporating network properties and molecular interactions into biomarker identification. For example, the MarkerPredict tool leverages network motifs and protein disorder to identify predictive biomarkers for cancer therapeutics. This approach achieved a remarkable LOOCV accuracy of 0.7-0.96 across 32 different models by accounting for the complex positioning of biomarkers within signaling networks [27]. Such performance demonstrates the advantage of systems-level thinking for clinical applications where biological complexity cannot be simplified.
Multi-Modal Data Integration Challenges
Biomedical research generates data across multiple biological layers and technological platforms, creating substantial integration barriers. Genomic, transcriptomic, proteomic, and metabolomic data each possess distinct characteristics, measurement scales, and noise profiles [72] [73]. This multi-modal heterogeneity is particularly problematic in systems biology, where the value emerges from integrating these diverse data streams to construct comprehensive network models.
The Alzheimer's disease research field exemplifies these challenges and opportunities. Multi-omics studies integrate data from genomics, epigenomics, transcriptomics, proteomics, lipidomics, and metabolomics to unravel the complex pathophysiology of neurodegeneration [73]. Each data type provides a partial view of the disease process, but only through integration can researchers identify coherent biomarker signatures with predictive power across biological scales. Successful integration requires sophisticated computational methods and standardized protocols to ensure compatibility between data types [72].
Technical and Analytical Variability
Beyond biological complexity, technical artifacts introduce significant variability that can obscure true signals. Batch effects - technical variations introduced during different experimental runs - pervade almost all high-throughput data [69]. These artifacts can lead to false discoveries if not properly accounted for in experimental design and statistical analysis. Studies have shown that technical errors can be mitigated through systematic data collection with standardized protocols, but complete elimination is often impossible [69].
Measurement variability extends to biomarker assessment methodologies. In wastewater-based epidemiology, classification models for C-Reactive Protein (CRP) concentrations achieved accuracies of only 64.88% to 65.48% despite using advanced machine learning algorithms [74]. This performance ceiling reflects the substantial technical noise inherent in complex sample matrices. Similarly, in clinical trials for Alzheimer's disease, plasma biomarkers exhibit both between-subject and within-individual variability that must be addressed through repeated measurements and specialized statistical designs [75].
Standardization Frameworks and Methodologies
Experimental Design Considerations
Robust biomarker research begins with rigorous experimental design that anticipates and controls for sources of variability. The SLIM design (Single-arm Lead-In with Multiple Measures) represents an innovative approach specifically developed to address variability challenges in early-phase clinical trials [75]. This design incorporates repeated biomarker assessments over short follow-up periods during both placebo lead-in and active treatment phases, improving measurement precision and statistical power while minimizing between-subject variability.
Table 2: Key Research Reagent Solutions for Biomarker Studies
| Reagent/Material | Function in Biomarker Research | Considerations for Standardization |
|---|---|---|
| Next-Generation Sequencing Kits | Genomic and transcriptomic profiling | Standardized library preparation protocols and quality control metrics [72] [73] |
| Protein Assay Panels | Multiplexed protein biomarker quantification | Calibration against reference standards, validation of cross-reactivity [76] |
| Liquid Biopsy Collection Tubes | Stabilization of circulating biomarkers | Pre-analytical variables including processing time and temperature [76] |
| Data Harmonization Tools | Integration of multi-omics datasets | Implementation of common data models and ontologies [69] |
| AI/ML Training Datasets | Model development and validation | Application of FAIR principles; comprehensive metadata [71] [70] |
Data Management and FAIR Principles
The FAIR Guiding Principles (Findable, Accessible, Interoperable, and Reusable) provide a critical framework for addressing data heterogeneity challenges in biomarker research [69] [70]. These principles emphasize the importance of rich metadata, standardized formats, and persistent identifiers to enhance data usability across research teams and projects. Implementing FAIR principles requires significant investment in data infrastructure and researcher training, but the long-term benefits include improved reproducibility and more efficient resource utilization.
Data harmonization - the process of aligning data from different sources to ensure consistency and compatibility - represents a particular challenge in systems biology approaches [69]. This process is supported by community standards, ontologies, and innovative automated systems. Biomedical research communities often define standardized ontologies to categorize and encode terminologies into a common language, facilitating harmonization across studies and institutions [69]. These efforts enable the integration of data across resources, allowing researchers to combine and compare datasets for more powerful analyses.
Diagram Title: Data Standardization Workflow for Systems Biology
Reproducibility Challenges in Complex Biomarker Studies
Analytical and Computational Reproducibility
The computational complexity of systems biology approaches introduces significant reproducibility challenges that extend beyond traditional wet-lab methodologies. AI and machine learning models for biomarker discovery are particularly vulnerable to reproducibility failures due to several factors: sensitive hyperparameter configurations, stochastic training processes, and data preprocessing variations [70]. These challenges are compounded by the common practice of inadequate documentation regarding model architecture, training procedures, and evaluation metrics.
A recent analysis of biomedical AI challenges revealed that 71% of researchers identified finding clean data as their primary hurdle, while 29% pointed to data annotation as the critical bottleneck [70]. This underscores the fundamental role of data quality in reproducible research. Unlike standardized datasets in other fields, biomedical data comes in multiple forms - microscopy images, genomic sequences, patient records - with no universal standard governing how these datasets should be stored, labeled, or structured [70]. This heterogeneity creates substantial barriers to reproducing published findings.
Strategies for Enhanced Reproducibility
Several promising strategies have emerged to address reproducibility challenges in complex biomarker research. Packaging research projects for reproducibility using containerization tools like Docker and code notebooks (Jupyter, R Markdown) helps capture the complete computational environment, including specific software versions and dependencies [71]. This approach ensures that analyses can be rerun consistently across different computing environments.
Meta-research (the study of research itself) provides another valuable approach for assessing and improving reproducibility. Quantitative meta-analysis integrates findings from multiple studies to reduce uncertainty and bias, though it requires careful handling of heterogeneity between studies [71]. When heterogeneity is present, appropriate statistical models must be employed to provide valid meta-analytic summaries that account for between-study differences.
Diagram Title: Multi-Layer Strategy for Research Reproducibility
Case Study: MarkerPredict - A Systems Approach to Predictive Biomarkers
Experimental Protocol and Methodology
The MarkerPredict framework exemplifies how systems biology principles can be applied to address data heterogeneity and reproducibility challenges in biomarker discovery [27]. This approach integrates network-based properties of proteins with structural features to identify predictive biomarkers for targeted cancer therapies. The experimental methodology can be summarized as follows:
Network Construction: Three distinct signaling networks with different topological characteristics were utilized: Human Cancer Signaling Network (CSN), SIGNOR, and ReactomeFI [27].
Motif Identification: Three-nodal motifs were identified using the FANMOD programme, with focus on fully connected triangles as regulatory hotspots in signaling networks [27].
Feature Integration: Network topological information was combined with protein annotations, including intrinsic disorder predictions from DisProt, AlphaFold, and IUPred databases [27].
Machine Learning Classification: Both Random Forest and XGBoost algorithms were trained on literature-evidence-based positive and negative training sets totaling 880 target-interacting protein pairs [27].
Validation: Model performance was evaluated using leave-one-out-cross-validation (LOOCV), k-fold cross-validation, and train-test splits (70:30) [27].
Biomarker Scoring: A Biomarker Probability Score (BPS) was defined as a normalized summative rank of the models to prioritize potential predictive biomarkers [27].
Performance Comparison with Traditional Approaches
The MarkerPredict framework demonstrates the power of systems biology to overcome limitations of reductionist approaches. The table below summarizes its performance compared to theoretical reductionist benchmarks:
Table 3: Performance Comparison of MarkerPredict vs. Theoretical Reductionist Benchmark
| Performance Metric | MarkerPredict (Systems Approach) | Theoretical Reductionist Benchmark |
|---|---|---|
| LOOCV Accuracy Range | 0.7 - 0.96 [27] | Not reported in search results |
| Number of Classified Pairs | 3,670 target-neighbor pairs [27] | Not reported in search results |
| Biomarkers Identified | 2,084 potential predictive biomarkers; 426 classified by all calculations [27] | Not reported in search results |
| Key Differentiating Features | Incorporates network motifs and protein disorder | Typically focuses on single molecular features |
| Clinical Relevance | High, due to systems-level context | Often limited by biological oversimplification |
This case study illustrates how embracing biological complexity through systems biology approaches can yield more robust and clinically relevant biomarkers compared to traditional reductionist methods. The integration of network properties with molecular features enables more accurate prediction of biomarker utility in heterogeneous patient populations.
The challenges of data heterogeneity, standardization, and reproducibility in biomarker research are substantial but not insurmountable. Addressing these issues requires a fundamental shift from reductionist to systems biology approaches that embrace rather than ignore biological complexity. This transition necessitates both conceptual and methodological innovations, including the development of standardized frameworks for data integration, robust computational pipelines for analysis, and comprehensive documentation practices for enhanced reproducibility.
The future of biomarker discovery lies in leveraging these systems-level approaches while maintaining rigorous attention to data quality and analytical transparency. As biomarker research increasingly incorporates artificial intelligence and multi-omics technologies, the principles outlined in this review will become even more critical for generating clinically meaningful findings. By adopting systems biology frameworks and addressing data challenges directly, researchers can unlock the full potential of biomarkers to guide personalized therapeutic strategies and improve patient outcomes across diverse disease contexts.
The field of biomarker research is undergoing a fundamental paradigm shift, moving from traditional reductionist approaches that study individual molecules in isolation toward systems biology frameworks that analyze complex biological networks as integrated wholes. Where reductionist methods have successfully identified single biomarkers like PSA for prostate cancer, they often suffer from limited diagnostic accuracy due to biological complexity [77]. Systems biology, by contrast, views biology as an information science and studies biological systems as complete entities interacting with their environment [77]. This approach recognizes that disease processes emerge from perturbations across interconnected molecular networks rather than isolated molecular defects. The computational modeling of these networks—from initial validation through dynamic simulation—represents both the greatest promise and most significant challenge in advancing predictive biomarker discovery for precision medicine.
This transition is driven by the recognition that disease-perturbed networks produce molecular fingerprints detectable well before clinical symptoms appear, offering unprecedented opportunities for early diagnosis and intervention [77]. However, capitalizing on this potential requires overcoming substantial computational challenges in model construction, validation, and simulation. This review examines these challenges through a comparative lens, evaluating traditional reductionist methodologies against emerging systems approaches, with particular focus on network validation techniques and dynamic simulation methodologies that are reshaping biomarker discovery and therapeutic development.
Comparative Framework: Systems Biology Versus Reductionist Biomarker Approaches
The fundamental distinction between systems biology and reductionist approaches lies in their conceptualization of biological systems and their strategies for biomarker discovery. Reductionist methods typically focus on linear causality and single-molecule biomarkers, while systems approaches employ network-level analyses that capture emergent properties and complex interactions [77] [78]. The following comparison outlines core methodological differences:
Table 1: Fundamental Differences Between Reductionist and Systems Biology Approaches
| Aspect | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Analytical Focus | Single molecules or pathways | Interacting networks and systems |
| Biomarker Strategy | Single biomarker identification | Multi-parameter molecular fingerprints |
| Causality Model | Linear causality | Network perturbations and emergent properties |
| Methodology | Hypothesis-driven | Data-driven and model-based |
| Temporal Dimension | Static measurements | Dynamic, time-resolved monitoring |
| Validation Criteria | Specificity and sensitivity for single marker | Network robustness and predictive accuracy |
| Diagnostic Application | Pauci-parameter diagnostics | Multi-parameter panel analyses |
| Therapeutic Implications | Single target drugs | Network-level interventions |
Reductionist approaches have demonstrated utility in identifying clinically relevant biomarkers, exemplified by prostate-specific antigen (PSA) for prostate cancer. However, their limitations include insufficient specificity and inability to capture disease heterogeneity [77]. Systems approaches address these limitations by analyzing dynamically changing networks that provide more comprehensive disease signatures. For example, systems analysis of prion disease identified 333 perturbed genes mapping onto four major protein networks that explained virtually every aspect of prion pathology, revealing new modules including iron homeostasis and leukocyte extravasation not previously associated with the disease [77].
The validation criteria differ substantially between these paradigms. Where reductionist methods emphasize specificity and sensitivity of individual markers, systems approaches evaluate network robustness, predictive accuracy, and dynamic stability. This shift requires increasingly sophisticated computational infrastructure capable of handling multi-omics data integration, with knowledge graphs recognized as essential for integrating and structuring disparate data sources [79].
Computational Challenges in Network Validation
Data Heterogeneity and Integration Barriers
Network validation in systems biology faces significant challenges stemming from data heterogeneity across multiple biological layers. Contemporary biomarker detection platforms—including single-cell sequencing, spatial transcriptomics, and high-throughput proteomics—generate comprehensive molecular profiles spanning genomic, transcriptomic, proteomic, and metabolomic dimensions [8]. Integrating these disparate data types with inconsistent ontologies and incomplete metadata remains a substantial bottleneck.
Researchers predominantly use public databases such as GeneBank and GISAID rather than relying solely on literature, yet issues with data quality, inconsistent ontologies, and lack of structured metadata often require retraining public models with proprietary data [79]. The academic community's reluctance to share raw data due to competitive concerns further exacerbates these challenges, creating significant obstacles to validation [79]. One participant in a computational biology roundtable noted: "This is a competitive area—even in academia. No one wants to publish and then get scooped. It's their bread and butter. The system is broken—that's why we don't have access to the raw data" [79].
Parameter Identifiability and Sensitivity Analysis
Parameter identifiability presents a fundamental challenge in network validation, particularly when separating kinetic parameters like rmax and KM (maximal enzymatic rate and enzymatic affinity) for incorporation of inter-individual variability [80]. The atorvastatin biotransformation model demonstrated how parameter sensitivity analysis under multiple experimental constraints significantly improves model validity [80].
This approach enables the creation of a consistent framework for precise computer-aided simulations in toxicology by systematically investigating parameter sensitivity and its impact on model verification, discrimination, and reduction [80]. The separation of rmax and KM parameters allows incorporation of separate information from pharmacokinetics and quantitative proteomics, facilitating the integration of regulatory networks responsible for variation in expression levels of enzymes, transporters, and receptors [80].
Table 2: Network Validation Challenges and Computational Solutions
| Validation Challenge | Computational Approach | Application Example |
|---|---|---|
| Data heterogeneity | Multi-modal data fusion | Knowledge graph integration [79] |
| Parameter identifiability | Sensitivity analysis | Atorvastatin biotransformation modeling [80] |
| Inter-individual variability | Population-scale modeling | Virtual liver populations [80] |
| Model reproducibility | Standardized governance protocols | Shared biomarker databases [8] |
| Network structure uncertainty | Module-based assembly | Bond graph frameworks [81] |
Modularity and Physical Consistency in Network Validation
Modular assembly approaches using bond graphs are emerging as powerful tools for ensuring physical consistency during network validation [81]. Bond graphs combine aspects of both modularity and physics-based modeling, applying principles from engineering to ensure biochemical models comply with fundamental conservation laws [81]. This approach enables large-scale models to be built from smaller submodules that communicate through clear and unambiguous interfaces while maintaining thermodynamic consistency [81].
The bond graph framework supports both computational modularity (the ability for models to communicate and interact in a physically consistent manner) and functional modularity (the ability of modules to be isolated from the effects of other modules) [81]. This is particularly valuable for validating network models against experimental data, as it ensures parameters remain biologically plausible throughout the validation process.
Dynamic Simulation Methodologies and Limitations
Deterministic Modeling of Biotransformation Processes
Dynamic simulation of biological networks requires sophisticated mathematical frameworks that capture temporal processes across multiple scales. The deterministic modeling of atorvastatin biotransformation exemplifies this approach, integrating comprehensive knowledge of metabolic and transport pathways with physicochemical properties [80]. This model comprised kinetics for transport processes and metabolic enzymes alongside population liver expression data, enabling assessment of the impact of inter-individual variability of concentrations of key proteins [80].
The atorvastatin model highlighted how dynamic simulations considering inter-individual variability of major enzymes (CYP3A4 and UGT1A3) based on quantitative protein expression data from a large human liver bank (n = 150) revealed significant variability in individual biotransformation profiles, underscoring the individuality of pharmacokinetics [80]. This approach demonstrated that predicting individual drug biotransformation capacity requires quantitative and detailed models that capture population-level diversity rather than idealized average behaviors.
Multi-Scale and Multi-omics Integration
Dynamic simulations increasingly incorporate multi-omics approaches that integrate genomics, proteomics, metabolomics, and transcriptomics to achieve a holistic understanding of disease mechanisms [13]. By 2025, this trend is expected to gain momentum, enabling identification of comprehensive biomarker signatures that reflect disease complexity [13]. The rise of multi-omics approaches represents a shift toward systems biology that promotes deeper understanding of how different biological pathways interact in health and disease [13].
The integration of single-cell analysis with multi-omics data provides a more comprehensive view of cellular mechanisms, paving the way for novel biomarker discovery [13]. Single-cell analysis technologies facilitate identification of rare cell populations that may drive disease progression or resistance to therapy, while simultaneously uncovering insights into tumor microenvironment heterogeneity [13]. These advances enable more targeted and effective interventions through improved dynamic simulation capabilities.
Artificial Intelligence-Enhanced Dynamic Simulation
Artificial intelligence and machine learning are revolutionizing dynamic simulation by enabling more sophisticated predictive models that forecast disease progression and treatment responses based on biomarker profiles [13]. AI-driven algorithms facilitate automated analysis of complex datasets, significantly reducing time required for biomarker discovery and validation [13]. By leveraging AI to analyze individual patient data alongside biomarker information, clinicians can develop tailored treatment plans that maximize efficacy while minimizing adverse effects [13].
However, AI-enhanced simulations face significant challenges, including data quality issues, model transparency, and regulatory compliance. Participants in computational biology roundtables have emphasized the need for AI outputs to include trust metrics, akin to statistical confidence scores, to assess reliability [79]. As one participant noted: "A trustworthiness metric would be highly useful. Papers often present conflicting or tentative claims, and it's not always clear whether those are supported by data or based on assumptions. Ideally, we'd have tools that can assess not only the trustworthiness of a paper, but the reliability of individual statements" [79].
Experimental Protocols for Model Validation and Simulation
Protocol 1: Weighted Gene Co-Expression Network Analysis (WGCNA) for Biomarker Identification
Purpose: Identify gene modules associated with clinical features and candidate biomarkers through systems biology approaches [78].
Methodology:
- Data Acquisition: Gene expression profiles are downloaded from databases such as Gene Expression Omnibus (GEO). For myocardial infarction and osteoarthritis studies, datasets may include GSE66360 (49 MI patients, 50 controls), GSE61144 (14 MI patients, 10 controls), GSE75181 (12 OA patients, 12 controls), and GSE55235 (10 OA patients, 10 controls) [78].
- Data Preprocessing: Normalize data and eliminate outlier samples by hierarchical clustering analysis. Set soft-thresholding power to 20 and correlation coefficient threshold to 0.9 [78].
- Network Construction: Establish adjacency matrix and hierarchical clustering to identify essential modules. Calculate correlation coefficients between modules and clinical characteristics separately for each condition [78].
- Module Selection: Select modules with high correlation coefficients for collecting candidate genes. Intersect results with differentially expressed genes (DEGs) analysis to identify common DEGs [78].
- Validation: Verify hub genes through Least Absolute Shrinkage and Selection Operator (LASSO) analysis, receiver operating characteristic (ROC) curves, and single-cell RNA sequencing analysis. Confirm differential expression in primary cells (e.g., cardiomyocytes, chondrocytes) using RT-qPCR [78].
Applications: This protocol successfully identified DUSP1, FOS, and THBS1 as shared biomarkers for myocardial infarction and osteoarthritis, revealing inflammation and immunity as common pathogenic mechanisms with MAPK signaling pathway playing a key role in both disorders [78].
Protocol 2: Dynamic Biotransformation Modeling with Population Variability
Purpose: Develop deterministic models of drug biotransformation that incorporate inter-individual variability of key enzymes [80].
Methodology:
- System Characterization: Comprehensive literature review of involved metabolic and transport pathways alongside physicochemical properties. For atorvastatin, this includes metabolic enzymes (CYP3A4, UGT1A3) and transport processes [80].
- Experimental Data Collection: Conduct time-series experiments using primary human hepatocytes cultured on collagen gel precoated plates at density of 1.5·106 cells/well. Incubate with compound (e.g., 10 μM atorvastatin) and collect samples at specified time-points for extracellular and intracellular metabolite measurement [80].
- Model Parameterization: Evaluate model on primary human hepatocytes and perform parameter identifiability analysis under multiple experimental constraints. Incorporate quantitative protein expression data from human liver banks (n = 150) [80].
- Sensitivity Analysis: Apply computational tools for parameter sensitivity analysis to improve model validity. Separate rmax and KM values to incorporate variability information [80].
- Dynamic Simulation: Perform simulations considering inter-individual variability of key enzymes based on quantitative protein expression data. Analyze variability in individual biotransformation profiles [80].
Applications: This approach created a consistent framework for precise computer-aided simulations in toxicology, highlighting individuality of pharmacokinetics and enabling prediction of individual drug biotransformation capacity [80].
Protocol 3: Modular Model Assembly Using Bond Graphs
Purpose: Construct large-scale dynamic models in systems biology using modular, physically consistent components [81].
Methodology:
- Module Definition: Divide biological system into manageable submodels that retain their identity while interacting with other system components. Implement "white-box" modularity allowing individual variables and components to be exposed as required [81].
- Bond Graph Implementation: Use bond graphs to combine aspects of modularity and physics-based modeling. Ensure connections between models comply with physical conservation laws [81].
- Module Interconnection: Connect submodels through clear and unambiguous interfaces defined using physical conservation laws. Maintain thermodynamic consistency across modules [81].
- Model Testing: Develop, test, and validate modules in isolation before incorporating into larger models. Use abstraction to instantiate multiple copies of repeated motifs [81].
- Granularity Adjustment: Swap submodels for alternative models with different levels of granularity for benchmarking and comparison. Track model provenance to enable incremental changes based on new measurements [81].
Applications: This protocol has been successfully applied to models of mitogen-activated protein kinase (MAPK) cascades to illustrate module reusability and glycolysis pathways to demonstrate granularity modification [81].
Visualization of Computational and Modeling Workflows
Systems Biology Biomarker Discovery Pipeline
Systems Biology Biomarker Discovery Pipeline: This workflow illustrates the sequential process from multi-source data acquisition through dynamic simulation to biomarker identification.
Network Validation and Simulation Architecture
Network Validation and Simulation Architecture: This diagram illustrates the iterative process of model development while highlighting key computational challenges at each stage.
Table 3: Essential Research Reagents and Computational Resources for Systems Modeling
| Resource Category | Specific Tools/Reagents | Function and Application |
|---|---|---|
| Biological Data Resources | Gene Expression Omnibus (GEO) [78] | Public repository of functional genomics data |
| CTD, GeneCards, DisGeNET [78] | Disease-gene association databases | |
| ENCODE and GENCODE [82] | Reference data for comparison and meta-analysis | |
| Computational Frameworks | Bond Graphs [81] | Physics-based modular model assembly |
| WGCNA [78] | Weighted gene co-expression network analysis | |
| LASSO Analysis [78] | Feature selection for high-dimensional data | |
| Analytical Platforms | Limma Package (R) [78] | Differential expression analysis |
| Digital Science Portfolio [79] | Literature review and knowledge graph tools | |
| Metaphacts [79] | Ontology-based semantic indexing | |
| Experimental Systems | Primary Human Hepatocytes [80] | Physiologically relevant metabolism models |
| Collagen Gel Precoated Plates [80] | Hepatocyte culture substrate | |
| Williams Medium E [80] | Serum-free hepatocyte culture medium | |
| Validation Tools | Single-cell RNA Sequencing [78] | Cellular resolution transcriptome validation |
| RT-qPCR [78] | Targeted gene expression confirmation | |
| ROC Curve Analysis [78] | Diagnostic performance assessment |
The field of computational biomarker research stands at a transformative juncture, where the integration of systems biology approaches with advanced modeling methodologies is overcoming traditional reductionist limitations. The challenges of network validation and dynamic simulation—while substantial—are being addressed through innovative computational frameworks that incorporate multi-omics data, population variability, and physical constraints. The emerging paradigm leverages AI-enhanced predictive analytics, multi-omics integration, and modular physically-consistent modeling to develop biomarker signatures that accurately reflect disease complexity.
As these computational approaches mature, they are increasingly being translated into clinical applications through liquid biopsy technologies, patient-centric biomarker panels, and real-time monitoring systems [13]. The continued development of standardized protocols, shared data resources, and validation frameworks will be essential for realizing the full potential of systems biology approaches in clinical practice. By effectively connecting biomarker discovery with practical clinical utilization, these integrated computational and experimental approaches offer a pathway toward truly personalized medicine based on comprehensive understanding of individual disease networks and dynamics.
The transition from promising preclinical discoveries to clinically useful biomarkers remains a significant hurdle in modern drug development. Despite remarkable advances in biomarker discovery, a troubling chasm persists, with less than 1% of published cancer biomarkers ultimately entering clinical practice [83]. This translational gap represents not only delayed treatments for patients but also substantial wasted investments and reduced confidence in biomarker research. The fundamental challenge lies in the tension between two competing approaches: reductionist methods that focus on single targets within isolated pathways, and systems biology frameworks that seek to understand biomarkers within the complex, interconnected networks that define living systems [1].
Reductionist approaches have historically dominated biomedical research, successfully identifying singular molecular entities with diagnostic or prognostic value. However, this methodology often fails to account for the complex, multi-scale interactions within biological systems, leading to promising preclinical biomarkers that prove inadequate in heterogeneous patient populations [1] [83]. In contrast, systems biology employs computational and mathematical methods to study complex interactions within biological systems, positioning it as a transformative discipline for biomarker development [1]. By mapping the intricate relationships between multiple molecular components and their phenotypic manifestations, systems biology offers a pathway to biomarkers that better reflect the complexity of human disease.
This review compares these competing paradigms through the lens of translational success, examining specific technologies, experimental methodologies, and validation frameworks that are bridging the gap between network models and clinically actionable biomarkers.
Comparative Analysis of Biomarker Development Approaches
The table below summarizes the core differences between traditional reductionist and systems biology approaches to biomarker development, highlighting their distinct methodologies, strengths, and limitations.
Table 1: Comparison of Reductionist versus Systems Biology Approaches in Biomarker Development
| Aspect | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Philosophical Basis | Studies components in isolation; "single-target" focus | Analyzes systems as integrated networks; multi-target focus |
| Typical Biomarker Type | Single molecules (genes, proteins, metabolites) | Biomarker signatures, network states, dynamic patterns |
| Experimental Design | Controlled conditions; uniform models | Heterogeneous samples; human-relevant models |
| Data Integration | Limited modalities; single-omics common | Multi-omics integration (genomics, transcriptomics, proteomics, metabolomics) |
| Translational Success Rate | Low (<1% of published biomarkers enter practice) [83] | Emerging evidence of improved prediction |
| Strengths | Simplified validation; clear mechanistic hypotheses | Captures biological complexity; identifies emergent properties |
| Limitations | Poor performance in heterogeneous human populations | Computational complexity; requires specialized expertise |
Systems Biology Technologies for Advanced Biomarker Discovery
Dynamic Network Biomarker (DNB) Platforms
Conventional static biomarkers capture molecular states at single time points, but dynamic network biomarkers (DNBs) monitor changes in regulatory networks across disease states, offering superior potential for tracking disease progression and therapeutic response. The TransMarker framework represents a cutting-edge approach to DNB identification, specifically designed to detect genes with shifting regulatory roles during disease progression [84].
The TransMarker methodology employs a sophisticated multi-stage process:
- Multilayer Network Construction: Encodes each disease state (e.g., normal, precancerous, metastatic) as a distinct layer in a multilayer graph, integrating prior protein-protein interaction data with state-specific gene expression from single-cell RNA sequencing.
- Graph Embedding: Uses Graph Attention Networks (GATs) to generate contextualized embeddings for each gene in each disease state, capturing both local and global topological features.
- Cross-State Alignment: Employs Gromov-Wasserstein optimal transport to quantify structural shifts in gene regulatory roles between disease states.
- Biomarker Prioritization: Ranks genes using a Dynamic Network Index (DNI) that captures significant regulatory variability, enabling identification of state-specific molecular switches [84].
Table 2: Performance Comparison of Network Biomarker Identification Methods
| Method | Network Type | Temporal Resolution | Validation Status | Reported Classification Accuracy |
|---|---|---|---|---|
| TransMarker [84] | Dynamic multilayer | Single-cell | Gastric cancer data | Superior to comparator methods |
| DyNDG [84] | Time-series multilayer | Bulk sequencing | Leukemia | Moderate |
| RL-GenRisk [84] | Static graph | Cross-sectional | Renal carcinoma | Moderate to high |
| Traditional Hub-Gene | Static network | Single time point | Various | Variable, often poor translation |
Multi-Omics Integration and Spatial Biology
Systems biology approaches increasingly rely on integrating multiple data modalities to capture biological complexity. Multi-omics profiling combines genomic, transcriptomic, proteomic, and metabolomic data to provide a holistic view of molecular processes, revealing biomarkers that might be missed when relying on a single data type [3]. For example, an integrated multi-omic approach played a central role in identifying the functional role of two genes, TRAF7 and KLF4, frequently mutated in meningioma [3].
Spatial biology technologies represent another advancement, preserving the architectural context of biomarkers within tissues. Techniques including spatial transcriptomics and multiplex immunohistochemistry (IHC) allow researchers to study gene and protein expression in situ without altering the spatial relationships between cells [3]. This spatial context is critical for biomarker identification, as the distribution of expression throughout a tumor often carries important biological information beyond mere presence or absence. For instance, studies suggest that the spatial distribution of immune cells within tumors can impact treatment response to immunotherapies [3].
Experimental Validation: From Discovery to Clinical Application
Machine Learning-Driven Biomarker Discovery
Artificial intelligence (AI) and machine learning are revolutionizing biomarker discovery by identifying subtle patterns in high-dimensional datasets that evade conventional analysis. These approaches are particularly valuable for integrating complex multi-modal data, including histopathology images, genomic profiles, and clinical information [7].
A representative example comes from liver fibrosis research, where researchers combined machine learning with experimental validation to identify neutrophil extracellular trap (NET)-associated biomarkers [85]. The experimental workflow included:
- Bioinformatic Analysis: Differential analysis and weighted gene co-expression network analysis (WGCNA) on GEO datasets (GSE84044, GSE49541) to identify NETs-related differentially expressed genes.
- Machine Learning Screening: Application of SVM-RFE and Boruta algorithms to prioritize biomarker candidates from 166 initial NETs-related genes.
- Experimental Validation: In vivo confirmation in a CCl4-induced murine fibrosis model, including immunohistochemistry, immunofluorescence, flow cytometry, and qPCR validation.
- Mechanistic Exploration: Identification of CCL2 as a key NETs-related liver fibrosis biomarker, along with its associated regulatory networks (miRNAs, lncRNAs) and potential therapeutic compounds [85].
This integrated computational-experimental approach demonstrates how machine learning can prioritize the most promising candidates from extensive molecular datasets before resource-intensive experimental validation.
Advanced Preclinical Models for Biomarker Validation
A significant limitation of traditional biomarker development has been the over-reliance on conventional animal models and 2D cell cultures with poor human correlation. Advanced models that better recapitulate human disease biology are now bridging this translational gap:
- Patient-derived organoids: 3D structures that recapitulate organ identity and retain characteristic biomarker expression patterns better than 2D cultures, enabling more accurate prediction of therapeutic responses [83].
- Patient-derived xenografts (PDX): Models derived from patient tumors and implanted into immunodeficient mice that better maintain the characteristics of human cancer, including tumor heterogeneity and evolution [83].
- Humanized mouse models: Systems that incorporate human immune components, allowing study of biomarker expression and therapeutic response in the context of human immune interactions [3].
- 3D co-culture systems: Platforms incorporating multiple cell types (immune, stromal, endothelial) to model the human tissue microenvironment more physiologically accurately [83].
These advanced models become particularly powerful when integrated with multi-omics technologies and longitudinal sampling strategies that capture temporal biomarker dynamics rather than single timepoint measurements [83].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Tools for Translational Biomarker Development
| Tool Category | Specific Technologies/Platforms | Key Applications | Considerations |
|---|---|---|---|
| Advanced Models | Patient-derived organoids, PDX, 3D co-culture systems | Biomarker validation, therapeutic response prediction | Better human correlation than traditional models |
| Spatial Biology | Spatial transcriptomics, multiplex IHC | Tissue context preservation, tumor microenvironment analysis | Reveals spatial biomarker patterns |
| Multi-Omics | Genomics, transcriptomics, proteomics, metabolomics | Comprehensive biomarker signatures, pathway analysis | Integrated analysis required for full potential |
| Computational Tools | TransMarker framework, AI/ML algorithms | Dynamic network biomarker identification, pattern recognition | Requires specialized bioinformatics expertise |
| Longitudinal Assays | Repeated plasma sampling, serial imaging | Capturing biomarker dynamics over time | More informative than single timepoints |
Visualizing Systems Biology Workflows
The following diagrams illustrate key computational and experimental workflows in systems biology-driven biomarker development.
Dynamic Network Biomarker Identification
Integrated Computational-Experimental Validation
The integration of systems biology approaches with advanced experimental models represents a paradigm shift in biomarker development, offering a promising path forward for bridging the translational gap. By moving beyond reductionist single-target approaches to embrace biological complexity, these integrated frameworks demonstrate improved capacity to identify biomarkers with genuine clinical utility. The convergence of multi-omics technologies, AI-driven analytics, human-relevant model systems, and dynamic network modeling is creating a new generation of biomarkers that more accurately reflect human disease complexity. As these approaches mature and standardization improves, they hold significant potential to transform biomarker development from a high-attrition endeavor to a more predictable, evidence-based process, ultimately accelerating the delivery of precision medicine to patients.
Complex diseases such as cancer, autism spectrum disorders, and coronary artery disease present a significant challenge for therapeutic development due to profound patient heterogeneity. This heterogeneity, stemming from diverse genetic, environmental, and molecular factors, results in variable treatment responses and has been a major contributor to high failure rates in clinical trials [86]. Traditional approaches to drug development have often relied on reductionist biomarker strategies, focusing on single molecules or linear pathways to identify patient subgroups. However, these methods frequently overlook the intricate biological networks that underlie disease mechanisms, limiting their ability to predict therapeutic response accurately [86] [30].
In contrast, systems biology approaches leverage holistic network analysis and multi-omics data integration to deconstruct this heterogeneity. By modeling the complex interplay of molecular components, these strategies can identify biologically coherent patient strata with distinct pathomechanisms and treatment response profiles [86] [87]. This guide provides a comparative analysis of these competing paradigms, examining their methodological foundations, performance characteristics, and utility for identifying responder subpopulations in drug development.
Comparative Analysis: Reductionist vs. Systems Biology Approaches
The following table summarizes the core distinctions between reductionist and systems biology approaches to patient stratification.
Table 1: Fundamental Comparison Between Stratification Approaches
| Feature | Reductionist Biomarker Approach | Systems Biology Approach |
|---|---|---|
| Philosophical Basis | Focuses on single biomarkers or linear pathways [30] | Holistic analysis of complex, interacting biological networks [86] [30] |
| Primary Objective | Identify single molecules (e.g., proteins, genes) with differential expression [88] | Identify differential network structures and interconnected molecular modules [86] [33] |
| Data Utilization | Typically analyzes one data type (e.g., genomics OR transcriptomics) | Integrates multi-omics data (genomics, transcriptomics, proteomics, clinical) [86] [87] |
| View of Heterogeneity | Often considered noise to be averaged out [86] | A core feature to be modeled and understood [86] |
| Patient Stratification | Based on individual biomarker thresholds (e.g., EGFR mutation status) [89] | Based on multivariate signatures, network perturbations, or pathway activities [86] [87] |
| Typical Output | A single predictive or prognostic biomarker (e.g., BRCA1 mutation) [89] | A patient-specific network or a stratification into distinct biotypes [86] [87] |
Performance Evaluation: Quantitative Outcomes and Experimental Data
When evaluated on key performance metrics, systems biology approaches demonstrate distinct advantages, particularly in managing complexity and biological interpretability.
Table 2: Performance Comparison for Patient Stratification
| Performance Metric | Reductionist Biomarker Approach | Systems Biology Approach |
|---|---|---|
| Accuracy in Heterogeneous Cohorts | Often limited; fails in diseases with multiple underlying causes [86] | Superior; identifies distinct biotypes within clinically homogeneous groups [87] |
| Biological Interpretability | Limited to a single molecule/pathway, often lacking mechanistic context [88] | High; embeds biomarkers within functional modules and pathways [86] [33] |
| Clinical Validation Success Rate | High attrition; many biomarkers fail to translate [90] | Emerging evidence suggests more robust translation [86] [87] |
| Ability to Discover Novel Biology | Low; constrained by pre-existing hypotheses | High; data-driven and capable of uncovering emergent properties [30] [88] |
| Handling of Genetic Complexity | Uses Polygenic Risk Scores (PRS), which are biologically agnostic [87] | Uses frameworks like CASTom-iGEx, which links liability to specific biological processes [87] |
A paradigmatic application of the systems approach, the CASTom-iGEx framework, demonstrated its superior capability in stratifying patients with Coronary Artery Disease (CAD). This method clusters patients based on tissue-specific imputed gene expression and pathway activity profiles, revealing biologically distinct subgroups that differed in intermediate phenotypes and clinical outcomes. Crucially, these clinically meaningful strata could not be identified using traditional PRS-based analysis, highlighting the limitation of the reductionist model [87].
Experimental Protocols and Methodologies
Protocol 1: Network-Based Biomarker Discovery for Colorectal Cancer
This protocol, derived from a published study, identifies diagnostic and prognostic biomarkers for colorectal cancer (CRC) using a systems biology workflow [33].
- Data Acquisition: Retrieve CRC gene expression datasets from public repositories like the Gene Expression Omnibus (GEO).
- Differential Expression Analysis: Identify Differentially Expressed Genes (DEGs) between tumor and normal tissues using tools such as R/Bioconductor.
- Network Reconstruction: Reconstruct a Protein-Protein Interaction (PPI) network using databases like STRING and visualize it with software such as Cytoscape or Gephi.
- Centrality Analysis: Analyze the PPI network to identify topologically central "hub" genes (e.g., based on degree, betweenness centrality). These hubs are potential key drivers of the pathology.
- Module Detection: Perform clustering analysis (e.g., using the k-mean algorithm) to dissect the PPI network into interactive modules or functional units.
- Enrichment Analysis: Conduct Gene Ontology (GO) and KEGG pathway enrichment analysis on the hub genes and modules to determine their biological functions and association with known disease pathways.
- Survival Analysis: Validate the prognostic value of the identified hub genes by performing survival analysis on independent cohorts using tools like GEPIA.
This workflow successfully identified 99 hub genes in CRC. It highlighted CCNA2, CD44, and ACAN as central to diagnosis and TUBA8, AMPD3, and TRPC1, among others, as linked to limited survival rates [33].
Protocol 2: A Multi-Objective Framework for Circulating miRNA Signatures
This protocol details a method to identify robust, functionally relevant circulating microRNA (miRNA) biomarkers for predicting colorectal cancer prognosis [88].
- Sample Collection and Preparation: Collect plasma from patients (e.g., with locally advanced or metastatic CRC) and healthy controls, followed by RNA isolation.
- Quality Control: Rigorously assess sample quality, for instance, by checking for haemolysis via free haemoglobin quantification and miR-16 levels.
- High-Throughput Profiling: Perform global miRNA profiling using a platform like OpenArray quantitative RT-PCR.
- Data Preprocessing: Preprocess the data (quality assessment, normalization, and missing data imputation) and dichotomize patients based on clinical outcome (e.g., long vs. short survival).
- Network Integration: Construct a knowledge-based miRNA-mediated gene regulatory network.
- Multi-Objective Optimization: Apply a computational framework that simultaneously optimizes for two objectives: the predictive power of the miRNA signature (based on expression data) and its functional relevance (based on the structure of the regulatory network).
- Validation: Confirm the altered expression of the identified miRNAs in an independent public dataset.
This integrative approach yielded a prognostic signature of 11 circulating miRNAs that reliably predicted patient survival and targeted pathways underlying CRC progression [88].
Visualization of Workflows and Signaling Pathways
The following diagram illustrates the core logical workflow of a systems biology approach to patient stratification, integrating multiple data types to define responder subpopulations.
The next diagram contrasts the fundamental logic of reductionist and systems-based approaches, highlighting their core differences in handling biological complexity.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of advanced patient stratification strategies requires a suite of specialized tools and reagents. The following table details key solutions for conducting these analyses.
Table 3: Essential Research Reagents and Tools for Patient Stratification
| Tool / Reagent | Function | Application Example |
|---|---|---|
| Cytoscape | Open-source software for visualizing and analyzing complex molecular interaction networks [30]. | Used to reconstruct and analyze PPI networks from DEGs to identify hub genes [33]. |
| STRING Database | A database of known and predicted protein-protein interactions, both physical and functional [30]. | Used to reconstruct the initial PPI network for a list of genes of interest (e.g., DEGs) [33]. |
| Patient-Derived Organoids | 3D in vitro models derived from patient tissues that recapitulate human tissue biology [90]. | Used in preclinical biomarker discovery to study patient-specific drug responses and model disease mechanisms. |
| Liquid Biopsy Assays | Enable non-invasive detection of biomarkers, such as circulating tumor DNA (ctDNA), from blood [90]. | Used for clinical biomarker monitoring, prognosis, and detecting minimal residual disease (MRD). |
| GTEx Dataset | A public resource containing tissue-specific gene expression and regulation data from post-mortem donors [87]. | Serves as a reference to train models for imputing tissue-specific gene expression from genotype data. |
| Ingenuity Pathway Analysis (IPA) | Commercial software for the analysis, integration, and interpretation of omics data in the context of biological pathways [30]. | Used for pathway analysis and functional annotation of gene lists derived from experimental data. |
| PriLer/CASTom-iGEx | A computational framework for stratifying patients based on tissue-specific imputed gene expression [87]. | Used for unsupervised discovery of clinically relevant patient strata (biotypes) from genetic data. |
The limitations of reductionist biomarker approaches are increasingly evident in the face of profound patient heterogeneity. While these methods remain valuable for well-defined, monogenic drivers, they are often insufficient for complex, polygenic diseases. The evidence demonstrates that systems biology approaches, which leverage network analysis and multi-omics data integration, provide a more powerful and biologically interpretable framework for patient stratification [86] [87]. They enable the move from a "one-size-fits-all" treatment model to a "type to treat" paradigm, where patient subtyping technologies identify those most likely to respond to a specific intervention [91].
The future of optimized patient stratification lies in the fusion of these approaches, leveraging the precision of molecular biomarkers within the rich, functional context provided by systems-level models [86]. As regulatory science evolves to embrace these complex biomarkers, the integration of systems biology into drug development holds the promise of derisking clinical programs and delivering more effective, personalized therapies to patients who need them most [89] [92].
The field of biological research is undergoing a fundamental transformation, moving away from traditional reductionist approaches toward more holistic systems methodologies. Where reductionism focuses on dissecting biological systems into their constituent parts and studying them in isolation, systems biology recognizes that health and disease emerge from the dynamic interactions within complex biological networks [93]. This paradigm shift necessitates a corresponding evolution in research team structures and resource allocation. The reductionist approach, while valuable for understanding individual components, cannot comprehend the complexity of biological systems whose properties cannot be explained or predicted by studying individual components alone [93]. Systems biology operates on the premise that the individual components of biological systems—such as molecular pathways—never work alone but operate in highly structured and integrated biological networks [93]. Consequently, understanding health and disease requires analyzing the changing dynamics of these networks through interdisciplinary collaboration that integrates analyses across broadly disparate levels, from molecular to organismal, and from genetic to environmental [93].
The transition to systems research represents more than merely a philosophical change—it demands fundamentally different team structures, expertise combinations, and resource allocations. Where traditional research might succeed with specialists working within their disciplinary silos, effective systems research requires the integration of diverse expertise to navigate biology's incredible complexity and apply these insights to clinical medicine [93]. This article compares the resource and expertise requirements for building successful interdisciplinary teams for systems research, contrasting them with traditional reductionist approaches, and provides practical frameworks for assembling and supporting these teams effectively.
Comparative Analysis: Resource Allocation Across Research Paradigms
Quantitative Comparison of Research Approaches
Table 1: Comparative analysis of reductionist versus systems biology approaches
| Characteristic | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Primary Focus | Individual components (genes, proteins) studied in isolation [93] | Dynamic interactions within biological networks [93] |
| Team Composition | Specialists within disciplinary silos | Interdisciplinary teams integrating multiple fields [94] |
| Data Generation | Targeted analysis of specific molecules | High-throughput multi-omics measurements (genomics, proteomics, metabolomics) [95] [96] |
| Infrastructure Needs | Standard laboratory equipment | Multiplexing technologies, high-performance computing, specialized software [93] |
| Technical Expertise | Deep knowledge in specialized methodology | Cross-training in computational and biological domains [94] |
| Time Investment | Faster initial setup | Significant time required for team integration and data integration [97] |
| Analytical Approach | Hypothesis-driven experimentation | Data-driven modeling and simulation [95] |
Expertise Requirements for Modern Systems Research
Table 2: Core competencies required for interdisciplinary systems research teams
| Domain Expertise | Specific Skills/Knowledge | Role in Systems Research |
|---|---|---|
| Biology/Immunology | Knowledge of specific biological systems, pathways, and disease mechanisms [95] | Provides fundamental biological context and insight into systems being studied [94] |
| Computational Biology | Data analysis, algorithm development, statistical modeling [95] [94] | Analyzes and interprets complex multi-omics datasets to extract biological meaning [94] |
| Bioinformatics | Programming, database management, tool development [95] | Develops and maintains computational infrastructure and analytical pipelines |
| Mathematics/Statistics | Mathematical modeling, network theory, dynamical systems [95] | Develops quantitative models of biological systems and their dynamics |
| Engineering | Technology development, instrumentation, optimization [94] | Designs and implements novel high-throughput measurement technologies |
| Data Visualization | Information design, visual analytics, visualization tools [98] | Creates intuitive visual representations of complex biological data and networks |
Methodological Framework: Implementing Interdisciplinary Systems Research
Experimental Design and Team Integration
Successful systems research requires methodological approaches that span traditional disciplinary boundaries. The workflow typically integrates both experimental and computational components in an iterative cycle of hypothesis generation, testing, and model refinement. A representative example can be found in systems immunology research, which combines multi-omics data, mechanistic models, and artificial intelligence to reveal emergent behaviors of immune networks [95]. These approaches leverage high-dimensional datasets including transcriptomics, proteomics, and metabolomics to develop predictive models of immune function and dysfunction [95].
Key Methodological Components:
Multi-omics Data Integration: Combining measurements across genomic, transcriptomic, proteomic, and metabolomic levels to capture system-wide dynamics [95] [96]
Network Analysis: Mapping molecular components and their interactions into structured networks to identify emergent properties [95]
Computational Modeling: Developing quantitative models that simulate system behavior under different conditions [95]
Experimental Validation: Testing model predictions using targeted experiments to refine understanding [95]
The integration of single-cell technologies—including scRNA-seq, CyTOF, and single-cell ATAC-seq—has been particularly transformative for systems immunology, revealing rare cell states and resolving heterogeneity that bulk omics approaches overlook [95]. These technologies provide high-dimensional inputs for data analysis, enabling cell-state classification, trajectory inference, and the parameterization of mechanistic models with unprecedented biological resolution [95].
Interdisciplinary Research Workflow
Diagram 1: Integrated interdisciplinary research workflow showing the iterative collaboration between computational and experimental domains.
Barrier Analysis: Challenges in Interdisciplinary Team Building
Structural and Attitudinal Barriers
Building successful interdisciplinary teams faces significant barriers that must be systematically addressed. These challenges can be categorized into five major areas: attitude, communication, academic structure, funding, and career development [97]. Despite widespread recognition of the need for interdisciplinary research, many scientists remain reluctant to abandon their disciplinary focus, with some viewing interdisciplinary science as "second-rate" or "less challenging" [97]. This attitudinal resistance often stems from concerns that those who engage in collaborative work cannot succeed in their own discipline or may "lose their professional identity" in team efforts [97].
Communication barriers present equally significant challenges, beginning with disciplinary jargon that creates misunderstandings between specialists from different fields [97]. The problem is compounded when the same terms have different meanings across disciplines, leading to situations where "different disciplines are continually rediscovering one another's discoveries, because they all have different names for them" [97]. Effective interdisciplinary collaboration requires substantial time and effort to learn the language of other fields and teach others the language of one's own discipline [97].
Institutional and Career Development Barriers
Traditional academic structures present formidable obstacles to interdisciplinary research. Most universities remain partitioned along academic lines that may no longer reflect today's intellectual frontiers, with these academic groupings serving primarily as categories for budgeting and administrative management [97]. The departmental structure of universities, which controls teaching, faculty recruitment, advancement, and promotion, changes relatively slowly and often fails to accommodate or reward interdisciplinary approaches [97].
Promotion and tenure policies represent particularly significant barriers, as these "major motivators and controlling devices for academic scientists" typically prioritize contributions within traditional departmental structures [97]. Junior faculty with interdisciplinary interests often face challenges in being viewed as making substantial contributions to their home departments, creating disincentives for pursuing systems approaches [97]. Additionally, institutional policies regarding allocation of laboratory space, hiring, and credit for successful grants frequently disadvantage researchers working across departmental boundaries [97].
Team Assembly Framework: Principles for Successful Collaboration
Strategic Team Composition and Leadership
Assembling effective interdisciplinary teams requires intentional strategies that address both technical and interpersonal dimensions. Successful teams blend diverse expertise while establishing clear principles for collaboration. Based on practical lessons learned from establishing multidisciplinary research teams, several key principles emerge [99]:
Table 3: Core principles for building and maintaining successful interdisciplinary research teams
| Principle | Implementation Strategies | Expected Outcomes |
|---|---|---|
| Clarify Roles & Expectations | Establish clear authorship policies, data sharing protocols, and resource allocation early in collaboration [97] [99] | Reduced conflicts, equitable credit distribution, efficient workflow |
| Foster Mutual Respect | Create opportunities for team members to appreciate the value and limitations of different methodologies [97] | Enhanced trust, willingness to integrate diverse perspectives |
| Develop Shared Language | Implement regular cross-training sessions, glossaries of terms, and structured communication formats [97] [94] | Reduced misunderstandings, more effective knowledge integration |
| Ensure Effective Leadership | Appoint mature scientists with established careers and experience in interdisciplinary research [97] | Better team coordination, navigation of institutional barriers |
| Build Trust Relationships | Facilitate informal interactions, team-building activities, and shared physical or virtual spaces [97] [94] | Stronger collaboration, increased information sharing |
Leadership selection critically influences interdisciplinary team success. Effective leaders must understand the challenges of group dynamics and possess the skills to establish and maintain an integrated program [97]. They need vision, creativity, and perseverance to educate scientific colleagues and administrators about the value of interdisciplinary research while coordinating the efforts of diverse team members [97]. Mature scientists with well-established research careers who have conducted interdisciplinary research of their own are often best positioned to direct these teams [97].
Physical and Virtual Collaboration Infrastructure
The design of collaboration environments significantly impacts interdisciplinary team effectiveness. Both physical spaces and digital infrastructure must facilitate communication and integration across disciplinary boundaries. Physical infrastructure considerations include:
- Open Laboratory Designs: Spaces with minimal physical boundaries that encourage spontaneous interactions and idea exchange between researchers from different backgrounds [94]
- Shared Common Areas: Central facilities and informal gathering spaces that promote chance interactions and scientific discussions [97]
- Co-location of Facilities: Strategic placement of core instrumentation at the center of work areas to increase interactions between technical experts and other researchers [94]
Virtual collaboration platforms are increasingly important for systems biology research. Systems like Kosmogora and ECellDive exemplify architectures designed to support collaboration in systems biology by ensuring biological data access, traceability, and integrity while providing immersive visualization capabilities [98]. These platforms address the challenge of biological data fragmentation across numerous databases by serving as centralized intermediaries that enable efficient querying and integration of diverse biological knowledge resources [98].
Essential Research Reagents and Computational Tools
Core Research Solutions for Systems Biology
Table 4: Essential research reagents and computational tools for interdisciplinary systems research
| Category | Specific Solutions | Function in Research |
|---|---|---|
| Multiplexing Technologies | Microarray analysis, multiplex qPCR, mass spectrometry, single-cell technologies (scRNA-seq, CyTOF) [95] [93] | Simultaneous measurement of hundreds to thousands of analytes for comprehensive system profiling |
| Computational Analysis Platforms | R/Bioconductor, Python computational libraries, specialized systems biology software [95] | Statistical analysis, data mining, and visualization of complex datasets |
| Data Management Systems | Kosmogora-like systems, biological databases (BioModels, MetaNetX, UniProt) [98] | Centralized access to biological knowledge, data traceability, and integrity maintenance |
| Modeling & Simulation Tools | Mechanistic modeling software, flux balance analysis, network analysis tools [95] [98] | Quantitative representation of biological systems and simulation of system dynamics |
| Visualization Applications | ECellDive, data visualization libraries, specialized VR applications [98] | Immersive exploration and interaction with biological data and models |
Implementation Framework for Collaborative Research
Diagram 2: Essential components for successful interdisciplinary systems research, integrating technical infrastructure, human expertise, and organizational support.
Training the Next Generation of Interdisciplinary Scientists
Formal and Informal Training Modalities
Developing effective interdisciplinary scientists requires innovative approaches that transcend traditional disciplinary training. Successful programs typically employ a combination of formal and informal training modalities to address the complex requirements imposed by the diversity of trainees [94]. Formal training includes structured coursework covering both the theory and practice of systems biology and its core technologies, such as gene expression technologies, proteomics, and data visualization/integration [94]. These courses provide a common experience and theoretical grounding that team members can reference when working collaboratively [94].
Informal training encompasses the extensive learning that occurs outside structured curricula and often proves most valuable for interdisciplinary development [94]. This flexible training approach provides individualized opportunities tailored to meet the needs of diverse trainees, facilitated by:
- Open Laboratory Designs: Physical spaces with minimal boundaries that encourage spontaneous exchanges between researchers from different scientific backgrounds [94]
- Centralized Core Facilities: Instrumentation spaces positioned at the center of work areas to increase interactions between technical experts and other researchers [94]
- Regular Research Retreats: Organization-wide events that allow everyone to share research updates with a broad audience, encouraging a culture of idea exchange [94]
- Interdisciplinary Discussion Groups: Flexible-format gatherings that alternate between journal clubs, topic debates, and methodological discussions [94]
Institutional Support Structures
Creating sustainable interdisciplinary training programs requires institutional commitment beyond individual research teams. Academic institutions must develop support structures that counter the traditional disciplinary biases in promotion, tenure, and resource allocation [97]. Successful models include:
- Interdisciplinary Programs and Centers: University-established entities that cross departmental boundaries to provide organizational homes for interdisciplinary work [97]
- Joint Appointment Mechanisms: Faculty positions that span multiple departments to facilitate cross-disciplinary collaboration and recognition [97]
- Revised Promotion Criteria: Tenure and advancement policies that value interdisciplinary contributions and team science [97]
- Seed Funding Programs: Targeted funding opportunities specifically for interdisciplinary initiatives that may face barriers in traditional grant mechanisms [97]
The Institute for Systems Biology (ISB) exemplifies a successful interdisciplinary training environment that unites diverse research programs under a common vision while allowing individuals to explore their specific research interests [94]. This organizational model blends aspects of goal-driven team science (characteristic of private industry) with the curiosity-driven research tradition of academia, creating a hybrid approach that maintains exploratory spirit while pursuing transformative medical applications [94].
Building successful interdisciplinary teams for systems research requires thoughtful integration of technical resources, human expertise, and organizational support. The transition from reductionist to systems approaches represents not merely a methodological shift but a fundamental transformation in how biological research is conceptualized, organized, and executed. Success depends on addressing the significant barriers to interdisciplinary collaboration while implementing proven principles for team assembly, leadership, and training.
Researchers and institutions that strategically invest in the necessary resources, expertise, and collaborative frameworks will be best positioned to advance our understanding of complex biological systems and translate these insights into clinical applications. By embracing the principles outlined in this comparison guide—including clear role definition, effective leadership, appropriate infrastructure, and innovative training—research teams can overcome traditional disciplinary boundaries and harness the full potential of systems approaches to address pressing challenges in biomedicine and therapeutic development.
Evidence and Efficacy: Comparative Analysis of Systems Biology Performance in Biomarker Development
The pursuit of reliable biomarkers for disease diagnosis and prognosis represents a critical frontier in modern medicine, yet this field is characterized by a fundamental methodological divide. On one side lies the established reductionist approach, which seeks to isolate and validate individual molecular markers through hypothesis-driven research. On the other stands the emerging systems biology paradigm, which employs computational and network-based analyses to identify multivariate biomarker signatures that reflect the complex interplay of biological systems [1] [100]. This paradigm clash is not merely philosophical; it has profound implications for diagnostic accuracy, prognostic reliability, and ultimately, clinical utility in patient care.
The reductionist approach, while responsible for many cornerstone biomarkers in clinical use today, faces significant challenges in the context of complex, multifactorial diseases. Single-target biomarkers often fail to capture disease heterogeneity and the intricate network of molecular interactions that drive pathology [1]. In contrast, systems biology approaches leverage high-throughput technologies and computational power to develop biomarker panels that can more comprehensively characterize disease states and predict clinical outcomes [100]. This comparative analysis objectively evaluates the performance characteristics of these competing methodologies across multiple dimensions, providing researchers and drug development professionals with evidence-based guidance for methodological selection in biomarker discovery and validation.
Performance Comparison: Quantitative Metrics Across Methodologies
Diagnostic Performance Metrics
Table 1: Comparison of Diagnostic Accuracy Metrics Across Methodological Approaches
| Methodology | Average Sensitivity | Average Specificity | Clinical Context | Evidence Strength |
|---|---|---|---|---|
| Single-Target Biomarkers (Reductionist) | Variable (0.65-0.85) | Variable (0.70-0.90) | Well-established for specific conditions (e.g., troponin for MI) | Multiple large validation studies [101] |
| Biomarker Panels (Systems) | Generally higher (0.75-0.95) | Generally higher (0.80-0.95) | Complex diseases (e.g., cancer, psychiatric disorders) | Growing evidence base [100] |
| Network-Based Signatures | Emerging data suggests superior performance | Emerging data suggests superior performance | Early-stage research across multiple disease areas | Limited but promising [88] |
Prognostic Performance Metrics
Table 2: Comparison of Prognostic Accuracy Metrics Across Methodological Approaches
| Methodology | Hazard Ratio Range | Concordance Index (Predictive Accuracy) | Feature Reduction Impact | Clinical Validation Stage |
|---|---|---|---|---|
| Clinical Parameters Alone | 1.5-2.5 | 0.60-0.65 | Not applicable | Established standard |
| Molecular Signatures (Systems) | 2.0-4.0 | 0.65-0.75 | Critical for performance | Progressive validation ongoing [102] |
| Integrated Clinical-Molecular | 2.5-5.0+ | 0.75-0.85 | Essential for model optimization | Limited examples available [88] |
The quantitative comparison reveals distinct performance patterns across methodological approaches. For diagnostic applications, biomarker panels derived from systems approaches generally demonstrate superior sensitivity and specificity compared to single-marker strategies, particularly for complex diseases like cancer and psychiatric disorders where multiple pathological processes converge [100]. The prognostic domain shows even more pronounced advantages for systems approaches, with multivariate signatures consistently outperforming conventional clinical parameters alone, as evidenced by higher hazard ratios and improved concordance indices in prediction models [88].
A critical factor in the performance of systems biology approaches is the method of feature reduction applied to high-dimensional data. Recent comparative evaluations indicate that knowledge-based feature transformation methods, particularly transcription factor activities and pathway activities, outperform both data-driven feature selection and simple gene expression markers for drug response prediction [102]. This finding underscores the value of incorporating biological prior knowledge into computational models, essentially bridging the gap between pure data-driven discovery and biologically-informed validation.
Experimental Protocols: Methodological Workflows in Practice
Reductionist Approach Protocol
The traditional reductionist methodology follows a linear, hypothesis-driven pathway with clearly defined stages:
Hypothesis Generation: Based on known biological pathways or preliminary data, a candidate biomarker is identified (e.g., a specific protein, gene, or metabolite).
Assay Development: Develop and optimize specific detection methods (e.g., ELISA for proteins, PCR for RNA) for accurate quantification of the candidate biomarker.
Sample Collection: Obtain relevant biological samples (tissue, blood, etc.) from well-characterized patient cohorts and control groups.
Measurement and Analysis: Quantify biomarker levels and establish correlation with clinical endpoints through statistical analysis.
Validation: Confirm findings in independent cohorts using the same standardized assay [101] [103].
This reductionist workflow emphasizes strict standardization, controlled variables, and incremental validation, making it particularly suitable for contexts where the underlying biology is well-understood and the disease mechanism can be attributed to specific molecular disruptions.
Systems Biology Approach Protocol
Systems biology employs an integrated, discovery-oriented workflow that embraces complexity:
Multi-Omics Data Generation: Simultaneously profile multiple molecular layers (genomics, transcriptomics, proteomics, metabolomics) from patient-derived samples.
Data Integration and Network Construction: Integrate diverse data types to construct molecular interaction networks relevant to the disease pathology.
Feature Selection/Reduction: Apply computational methods to identify the most informative biomarkers from high-dimensional data:
- Knowledge-based: Pathway activities, transcription factor activities, drug pathway genes
- Data-driven: Principal components, autoencoders, regularization methods [102]
Predictive Model Building: Develop multivariate models using machine learning algorithms (ridge regression, random forest, SVM, etc.) that integrate the selected features.
Validation and Iteration: Test model performance in independent datasets and refine based on biological plausibility and clinical relevance [1] [88].
This protocol emphasizes holistic analysis, pattern recognition, and computational modeling, making it particularly advantageous for complex diseases with heterogeneous underlying mechanisms.
Methodological Workflows Comparison: This diagram illustrates the fundamental differences between reductionist and systems biology approaches to biomarker discovery, highlighting the linear nature of reductionist methods versus the iterative, multi-dimensional nature of systems approaches.
Case Study: Circulating miRNA Biomarkers in Colorectal Cancer
A compelling illustration of the practical implementation of these methodologies comes from research on circulating microRNA (miRNA) biomarkers for colorectal cancer (CRC) prognosis. This example demonstrates how a systems biology approach can address the limitations of reductionist strategies in a clinically challenging context.
Experimental Protocol for Network-Based miRNA Discovery
Patient Cohort and Sample Collection: 97 patients with histologically confirmed locally advanced or metastatic CRC were enrolled prospectively. Plasma samples were collected prior to chemotherapy initiation using standardized protocols (EDTA tubes, centrifugation within 30 minutes, storage at -80°C) [88].
RNA Isolation and Quality Control: Total RNA was isolated from plasma using the MirVana PARIS miRNA isolation kit with modifications. Quality control assessments included haemolysis evaluation through free haemoglobin quantification and miR-16 level measurement to exclude compromised samples [88].
miRNA Profiling: Global miRNA profiling was performed using the OpenArray platform with quantitative RT-PCR. The platform enabled simultaneous measurement of 754 miRNAs in each plasma sample, generating high-dimensional molecular data [88].
Statistical Preprocessing and Normalization: Raw Cq values underwent rigorous preprocessing including quality assessment, quantile normalization, missing data imputation using KNNimpute, and filtering of miRNAs with >50% missing values across samples. Patients were dichotomized into long versus short survival groups using a 2-year cutoff [88].
Network-Enhanced Biomarker Discovery: The innovative multi-objective optimization framework integrated:
- miRNA expression data from plasma samples
- miRNA-mediated gene regulatory network knowledge
- Functional relevance and predictive power as dual optimization targets [88]
This integrated approach identified an 11-miRNA signature that significantly predicted patient survival outcomes and targeted pathways underlying colorectal cancer progression, with independent validation confirming altered expression of these miRNAs in early versus advanced stage disease [88].
CRC miRNA Discovery Workflow: This diagram outlines the integrated experimental and computational workflow for identifying network-based miRNA biomarkers for colorectal cancer prognosis, highlighting the combination of empirical data generation with prior biological knowledge.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for Biomarker Discovery Methodologies
| Reagent/Platform | Specific Function | Methodological Context | Key Characteristics |
|---|---|---|---|
| OpenArray miRNA Panels | High-throughput miRNA profiling | Systems Biology | Enables simultaneous quantification of 754 miRNAs via qRT-PCR [88] |
| MirVana PARIS Kit | RNA isolation from plasma/serum | Both Approaches | Specialized for miRNA recovery from biofluids; compatible with downstream applications [88] |
| LINCS L1000 Landmark Genes | Feature reduction for transcriptomics | Systems Biology | 978 genes capturing ~80% of transcriptomic information [102] |
| Reactome Pathway Database | Knowledge-based feature generation | Systems Biology | Curated pathway information for biological context interpretation [102] |
| OncoKB Curated Cancer Genes | Clinically relevant gene set | Both Approaches | Expert-curated resource of clinically actionable cancer genes [102] |
| QUADAS (Quality Assessment Tool) | Methodological quality assessment | Reductionist | Validated tool for quality appraisal of diagnostic accuracy studies [103] |
The research toolkit for biomarker discovery varies significantly between methodological approaches, reflecting their different underlying philosophies and technical requirements. Reductionist approaches rely heavily on targeted, highly specific reagents like ELISA kits and PCR assays that enable precise quantification of individual analytes. In contrast, systems biology approaches require platforms capable of generating high-dimensional data, such as the OpenArray system for miRNA profiling, coupled with computational resources for data integration and analysis [88].
A critical emerging trend is the development of resources that support knowledge-based feature reduction and interpretation. Databases like Reactome and OncoKB provide structured biological knowledge that can be integrated with empirical data to enhance the biological plausibility and clinical relevance of discovered biomarkers [102]. This hybrid approach represents the cutting edge of biomarker research, leveraging the strengths of both high-throughput data generation and curated biological knowledge.
Discussion: Integration as the Path Forward
The evidence compiled in this comparative analysis suggests that the dichotomy between reductionist and systems approaches may be counterproductive. Rather than representing mutually exclusive alternatives, these methodologies form a complementary continuum in biomarker research. The most promising path forward appears to be integrative approaches that combine the statistical power of high-dimensional data with the biological insight of prior knowledge [88].
Systems biology approaches demonstrate particular strength in the discovery phase, where their ability to identify multivariate signatures captures complex disease biology more effectively than single-marker strategies. This is especially valuable for complex diseases like cancer, psychiatric disorders, and autoimmune conditions, where disease heterogeneity and multifactorial etiology have historically hampered biomarker development [1] [100]. The documented superiority of knowledge-based feature reduction methods like transcription factor activities and pathway activities further underscores the value of integrating biological insight with data-driven discovery [102].
However, reductionist methodologies retain important advantages in validation and clinical implementation, where their focus on specific, well-characterized analytes facilitates assay standardization and regulatory approval. The practical reality is that systems-derived biomarker panels must eventually be translated into clinically implementable assays, often requiring simplification to the most informative components [100].
Future directions in biomarker research will likely focus on refining hybrid methodologies that maintain the discovery power of systems approaches while addressing the practical constraints of clinical implementation. This includes developing more sophisticated computational methods for feature reduction, establishing standards for validating multivariate signatures, and creating regulatory pathways for the clinical adoption of network-based biomarkers. As these methodological bridges continue to strengthen, the field moves closer to realizing the promise of precision medicine through biomarkers that truly reflect the complexity of human disease.
The field of biomarker discovery is undergoing a fundamental transformation, moving from traditional reductionist approaches to sophisticated systems biology frameworks. Reductionist methods have historically focused on isolating and studying single biomarkers—such as individual proteins or genetic mutations—within linear pathways. While this approach has produced valuable diagnostic tools, it often overlooks the complex, interconnected nature of biological systems, potentially missing crucial interactions that underlie disease pathology and treatment response [104]. In contrast, systems biology approaches leverage multi-omics data integration, advanced computational modeling, and network-based analyses to capture the full complexity of disease mechanisms [8]. This paradigm shift enables the identification of biomarker signatures that more accurately reflect disease heterogeneity and progression.
The validation pathway for systems-derived biomarkers presents unique challenges and requirements that differ substantially from traditional biomarker validation. It requires a rigorous, multi-stage process that moves from computational prediction to clinical confirmation, ensuring that these complex signatures provide reliable, actionable insights for patient care and drug development [105]. This guide provides a comprehensive comparison of the methodologies, experimental protocols, and analytical frameworks essential for robust validation of systems-derived biomarkers, offering researchers a structured pathway from discovery to clinical implementation.
Methodological Comparison: Systems Biology vs. Reductionist Approaches
The fundamental differences between systems biology and reductionist methodologies shape every stage of biomarker discovery and validation. The table below summarizes the core distinctions between these competing paradigms.
Table 1: Core Methodological Differences Between Systems Biology and Reductionist Approaches
| Aspect | Reductionist Approach | Systems Biology Approach |
|---|---|---|
| Philosophical Foundation | Studies components in isolation to understand a system | Studies interactions and networks within a system as a whole |
| Data Type | Single-omics, univariate analysis | Multi-omics integration (genomics, proteomics, metabolomics, etc.) |
| Primary Technology | ELISA, PCR, targeted sequencing | High-throughput sequencing, mass spectrometry, AI/ML platforms |
| Network Consideration | Minimal; focuses on linear pathways | Central; analyzes complex interactions and network motifs |
| Typical Output | Single biomarker or small panels | Multivariate biomarker signatures or complex molecular classifiers |
| Handling of Heterogeneity | Limited; often averages out biological noise | Integral; can model and stratify based on heterogeneity |
The systems biology framework is particularly powerful for identifying predictive biomarkers in complex diseases like cancer. For instance, the MarkerPredict tool utilizes network motifs and protein disorder characteristics to identify potential predictive biomarkers for targeted cancer therapies. By analyzing proteins within interconnected three-nodal motifs in signaling networks, this systems-based approach has classified thousands of target-neighbor pairs, identifying 426 high-probability predictive biomarkers across multiple cancer signaling networks [27]. This stands in stark contrast to traditional, reductionist methods that typically focus on single, pre-defined biomarkers based on existing scientific knowledge.
The Validation Workflow: From Computational Prediction to Clinical Confirmation
Validating systems-derived biomarkers requires a structured, multi-phase workflow that ensures both analytical robustness and clinical relevance. The following diagram illustrates this comprehensive pathway.
Diagram 1: Comprehensive Validation Workflow for Systems-Derived Biomarkers. This pathway illustrates the multi-stage process from initial discovery through to clinical implementation, highlighting both computational and experimental phases.
Stage 1: Computational Discovery & Prioritization
The initial discovery phase leverages high-throughput technologies and computational power to identify potential biomarker signatures from vast molecular datasets.
Multi-Omics Data Integration: Modern discovery integrates data from genomics, transcriptomics, proteomics, and metabolomics to build comprehensive molecular maps of disease processes. Platforms like Polly by Elucidata streamline this process by harmonizing diverse datasets, making them machine learning-ready and addressing a major bottleneck in biomarker discovery [104].
Network-Based Analysis: Systems approaches analyze biological data within the context of interaction networks. For example, examining network motifs—specific patterns of interconnections—can reveal functionally important relationships. Research shows that proteins within interconnected three-node motifs with drug targets are enriched for predictive biomarkers in oncology [27].
Machine Learning Prioritization: AI/ML algorithms are crucial for analyzing these complex, high-dimensional datasets. Random Forest and XGBoost models have demonstrated high accuracy (0.7-0.96 LOOCV accuracy) in classifying potential predictive biomarkers, enabling researchers to prioritize the most promising candidates for experimental validation [27].
Stage 2: Analytical Validation
Once candidate biomarkers are identified, they must undergo rigorous analytical validation to ensure reliable measurement.
Assay Development: Developing robust assays that can accurately measure the biomarker signature in clinically relevant samples. For complex signatures, this may require multiplex assays capable of simultaneously measuring multiple analytes.
Technical Performance Evaluation: Establishing key analytical performance metrics including sensitivity (true positive rate), specificity (true negative rate), precision, and reproducibility across different laboratory conditions and operators [105].
Reference Standard Correlation: Ensuring the new assay shows strong correlation with established reference methods where available, particularly when transitioning from discovery platforms (e.g., sequencing) to clinically implementable assays (e.g., PCR).
Stage 3: Clinical Validation
Clinical validation establishes whether the biomarker reliably predicts the clinical outcome of interest in the target population.
Retrospective Studies: Initially, biomarker performance is typically evaluated using archived specimens from previously conducted studies or clinical trials. Proper study design is critical, including randomization and blinding to prevent bias during specimen selection and analysis [105].
Prognostic vs. Predictive Differentiation: A crucial distinction must be made between prognostic biomarkers (which provide information about overall disease outcomes regardless of therapy) and predictive biomarkers (which inform treatment response). Predictive biomarkers require evidence of a significant interaction between the biomarker and treatment effect, ideally from randomized controlled trials [105].
Performance Metrics: Clinical validity is established through statistical measures including discrimination (ability to distinguish cases from controls, often measured by AUC), calibration (accuracy of risk estimates), and clinical validity (strength of association with the clinical endpoint) [105].
Stage 4: Clinical Utility & Implementation
The final stage establishes whether using the biomarker improves patient outcomes and is feasible in real-world settings.
Clinical Impact Assessment: Evaluating whether biomarker-guided decision-making leads to improved health outcomes, reduced side effects, or more efficient resource utilization compared to standard care.
Health Economic Analysis: Assessing cost-effectiveness and economic impact of implementing the biomarker testing strategy within the healthcare system.
Clinical Guideline Integration: Successful biomarkers are incorporated into professional treatment guidelines and standards of care, facilitating widespread adoption into clinical practice.
Case Study Comparisons: Experimental Protocols & Outcomes
Case Study 1: Alzheimer's Disease ATN Biomarkers
A systematic comparison of A/T/N (amyloid/tau/neurodegeneration) biomarkers in Alzheimer's disease provides a compelling example of systems-derived biomarker validation in neurodegenerative disease.
Table 2: Performance Comparison of Alzheimer's Disease Biomarkers for Tracking Cognitive Decline
| Biomarker | Modality | Association with Cognitive Decline | Advantages | Limitations |
|---|---|---|---|---|
| Amyloid-PET | Imaging | Not significant in longitudinal studies | Gold standard for Aβ target engagement | Plateaus early; poor tracker of short-term change |
| Tau-PET | Imaging | Strong correlation | Excellent tracking of disease-stage progression | High cost; limited accessibility |
| Plasma p-tau217 | Fluid biopsy | Strong correlation | High specificity for AD; cost-effective; accessible | Requires standardized assays |
| Cortical Thickness | MRI | Strong correlation | Widely available; strong correlation with cognition | Confounded by pseudo-atrophy in anti-Aβ treatment |
Experimental Protocol: The study analyzed longitudinal data from the Alzheimer's Disease Neuroimaging Initiative (ADNI, N=141) and the A4/LEARN studies (N=151). Participants underwent repeated biomarker assessments (amyloid-PET, tau-PET, plasma p-tau217, MRI) and cognitive testing (MMSE, ADAS13, CDR-SB, PACC). Linear mixed models estimated change rates for both biomarkers and cognition, with bootstrapping used to compare predictive strengths across biomarkers [106].
Key Findings: The research demonstrated that longitudinal changes in tau-PET, plasma p-tau217, and cortical thickness—but not amyloid-PET—effectively tracked cognitive decline. Plasma p-tau217 emerged as a robust, cost-effective alternative to tau-PET, offering similar predictive power with greater accessibility for clinical monitoring [106].
Case Study 2: Metastatic Colorectal Cancer (mCRC) Predictive Model
This study exemplifies the application of AI/ML for developing predictive biomarkers for therapy response in oncology.
Experimental Protocol:
- Sample Collection: Formalin-fixed paraffin-embedded (FFPE) tumor samples from mCRC patients collected before treatment initiation.
- Multi-Omics Profiling:
- Mutational profiling of 50 CRC-related genes using next-generation sequencing platforms.
- Whole-transcriptome analysis using Affymetrix HTA2.0 arrays.
- Chromosomal instability analysis using high-resolution SNP genotyping arrays.
- Model Development: Machine learning algorithms (Random Survival Forest, neural networks) trained on multi-omics data to predict response to chemotherapy ± targeted therapy.
- Validation: Internal validation through cross-validation followed by external validation using public datasets (TCGA, GEO) [107].
Key Outcomes: The AI-derived model achieved high discrimination in distinguishing responders from non-responders, with area under the curve (AUC) values of 0.90 in training and 0.83 in validation datasets. This demonstrates the potential of systems-based approaches to identify complex molecular signatures that predict treatment response more accurately than single biomarkers [107].
Essential Research Toolkit for Biomarker Validation
Successful validation of systems-derived biomarkers requires specialized reagents, technologies, and computational resources. The following table details key components of the research toolkit.
Table 3: Essential Research Toolkit for Systems-Derived Biomarker Validation
| Tool Category | Specific Technologies/Platforms | Primary Function | Key Considerations |
|---|---|---|---|
| Multi-Omics Platforms | LC-MS/MS, GC-MS, NMR, RNA-seq, ATAC-seq | Comprehensive molecular profiling across biological layers | Platform compatibility, batch effect correction |
| Bioinformatics Solutions | Polly, MarkerPredict, custom Python/R pipelines | Data harmonization, machine learning, network analysis | FAIR compliance, reproducibility, scalability |
| AI/ML Frameworks | Random Forest, XGBoost, Neural Networks | Pattern recognition, biomarker prioritization, prediction | Interpretability, hyperparameter optimization |
| Validation Assays | Multiplex immunoassays, ddPCR, NGS panels | Translating discoveries to clinically applicable tests | Sensitivity, specificity, reproducibility |
| Data Management | LIMS, eQMS, EHR integration systems | Ensuring data integrity, traceability, and compliance | Interoperability, security, regulatory alignment |
The integration of these tools into a cohesive workflow is critical for efficient biomarker validation. Platforms that enable multi-omics integration and provide ML-ready data—such as Polly, which accelerated biomarker discovery timelines by sevenfold in one case study—demonstrate the practical impact of optimized toolkits [104].
Challenges and Future Directions
Despite significant advances, several challenges remain in the validation and implementation of systems-derived biomarkers.
Data Heterogeneity and Standardization: Integrating diverse data types from multiple sources remains a substantial obstacle. Variations in sample collection, processing protocols, and analytical platforms can introduce biases that compromise biomarker performance [8]. Solutions include implementing standardized governance protocols and adopting FAIR (Findable, Accessible, Interoperable, and Reusable) data principles [104].
Model Generalizability: Many biomarker models demonstrate excellent performance in discovery cohorts but fail to maintain accuracy in diverse, independent populations. This challenge requires intentional inclusion of diverse patient populations in training datasets and rigorous external validation across multiple clinical sites [8].
Regulatory Adaptation: Current regulatory frameworks for biomarker approval are evolving to accommodate complex, algorithm-based signatures. The European Union's In Vitro Diagnostic Regulation (IVDR) exemplifies both the progress and challenges in this area, with increasing recognition of real-world evidence but also creating uncertainty through inconsistent implementation across jurisdictions [6].
Clinical Translation Barriers: Even after robust validation, integrating systems-derived biomarkers into clinical workflows faces practical obstacles including physician acceptance, workflow integration, and reimbursement structures. Successful implementation requires close collaboration between researchers, clinicians, and healthcare systems from early development stages [6].
Future innovation will likely focus on dynamic biomarker monitoring through wearable sensors and liquid biopsies, advanced AI architectures for improved pattern recognition, and edge computing solutions for implementation in low-resource settings [13]. As these technologies mature, they will further accelerate the transition from reductionist to systems-based approaches in biomarker development, ultimately enabling more precise, personalized, and proactive healthcare.
The discovery of biomarkers, objectively measurable indicators of biological processes, has traditionally followed a reductionist paradigm, focusing on identifying single molecules with diagnostic or predictive value [8]. This approach, successful for some monogenic disorders, faces significant challenges in complex diseases like cancer and neurological disorders, where phenotypic outcomes arise from intricate interactions between genetic, environmental, and immunological factors [10]. Systems biology has emerged as a complementary field, shifting focus from isolated components to the interactions within complex networks [10]. This paradigm shift underpins the development of network biomarkers, which leverage relationships between molecules, and dynamic network biomarkers (DNBs), which capture temporal fluctuations to detect critical transitions in disease states [108]. This guide objectively compares the specificity and robustness of these systems-level biomarkers against traditional single-molecule markers, providing researchers and drug development professionals with a framework for selecting appropriate methodologies based on research and clinical goals.
Theoretical Foundations: Defining the Biomarker Classes
Single-Molecule Markers
Single-molecule markers are defined by the differential expression or concentration of individual molecules (e.g., genes, proteins, metabolites) between distinct states, such as health and disease [108]. Their discovery is typically hypothesis-driven, originating from known pathways, and their validation relies on establishing a statistically significant association between the molecule's level and a specific clinical outcome.
Network Biomarkers
Network biomarkers move beyond individual molecules to utilize the differential associations or correlations between pairs of molecules [108]. They are founded on the principle that diseases often arise from perturbations in biological networks rather than alterations in a single component. By capturing the interactions between molecules, they reflect the underlying system's stability and functional state.
Dynamic Network Biomarkers (DNBs)
DNBs represent a further evolution, designed to detect pre-disease states or critical tipping points before a system transitions into a manifest disease state [108]. They are characterized by the differential fluctuations and correlations within a group of molecules, signaling a loss of system resilience and an imminent phase transition. This makes them uniquely powerful for predictive and preventative medicine.
The conceptual relationships and evolution of these biomarker types are illustrated below.
Comparative Performance Analysis: Specificity and Robustness in Focus
The following tables synthesize quantitative and qualitative data from key studies to compare the performance of the three biomarker classes across critical metrics.
Table 1: Comparative Analysis of Specificity and Diagnostic Power
| Performance Metric | Single-Molecule Markers | Network Biomarkers | Dynamic Network Biomarkers (DNBs) |
|---|---|---|---|
| Diagnostic Specificity | Limited; often confounded by heterogeneity [8]. | Higher; captures context-specific network rewiring [84]. | Designed for pre-disease state specificity; detects imminent transitions [108]. |
| Biological Insight | Isolated; identifies "what" is altered but not "how" or "why" [10]. | Pathway-level; reveals "how" molecules interact in a disease state [84]. | System-level; reveals "why" a system becomes unstable before a critical shift [108]. |
| State Discrimination | Distinguishes disease from normal states. | Distinguishes disease subtypes and molecular contexts [84]. | Identifies pre-disease state, critical transition state, and normal state [108]. |
| Representative Experimental Finding | A specific gene mutation may be present in only a subset of patients, limiting its diagnostic coverage [84]. | The TransMarker framework achieved superior classification of gastric adenocarcinoma states by analyzing network rewiring [84]. | DNBs can provide an early-warning signal for a disease, enabling preventative intervention before symptom onset [108]. |
Table 2: Comparative Analysis of Robustness and Translational Potential
| Performance Metric | Single-Molecule Markers | Network Biomarkers | Dynamic Network Biomarkers (DNBs) |
|---|---|---|---|
| Robustness to Noise | Low; individual molecule measurements are susceptible to technical and biological variance [8]. | Higher; network structures are more stable as they are defined by multiple relationships [108]. | High; relies on collective fluctuation patterns, which are robust to minor individual variations. |
| Generalizability | Often poor across diverse populations due to genetic and environmental heterogeneity [8]. | Improved; network structures can be more conserved than individual marker levels [108]. | Context-dependent; generalizability of a specific DNB requires validation across cohorts. |
| Clinical Application | Well-established in current diagnostics (e.g., PSA testing). | Emerging role in precision oncology for patient stratification and drug response prediction [27]. | Primarily in research; holds potential for predictive medicine and forecasting disease flares. |
| Key Limitation | High false-negative/false-positive rates in complex diseases; misses compensatory mechanisms [108]. | Computationally intensive; requires high-quality interaction data; complex interpretation [84]. | Requires dense longitudinal data; identification of critical state window is challenging [108]. |
Experimental Protocols and Workflows
Protocol for Identifying a Single-Molecule Marker
This protocol outlines the standard workflow for a differential expression analysis.
- Sample Collection: Obtain biological samples (e.g., tissue, blood) from well-defined cohorts (e.g., disease vs. healthy control).
- Molecular Profiling: Use a high-throughput technology (e.g., RNA sequencing, mass spectrometry-based proteomics) to quantify molecule abundance in all samples.
- Data Preprocessing: Perform quality control, normalization, and batch effect correction on the raw data.
- Statistical Analysis: Apply hypothesis tests (e.g., t-test, ANOVA) to identify molecules with statistically significant differential abundance between cohorts. Correct for multiple hypothesis testing (e.g., using False Discovery Rate).
- Validation: Confirm the candidate marker using an independent technical method (e.g., qPCR) and/or in an independent validation cohort.
Protocol for Constructing a Network Biomarker: The TransMarker Workflow
The TransMarker framework is a modern method for identifying dynamic network biomarkers in cancer progression using single-cell data [84]. The detailed workflow is as follows:
- Multi-State Data Input: Collect single-cell RNA-sequencing data from multiple disease states (e.g., normal, pre-cancer, tumor).
- Multilayer Network Construction: Encode each disease state as a distinct layer in a multilayer graph. For each layer:
- Integrate prior knowledge of gene-gene interactions (e.g., from protein-protein interaction databases).
- Use state-specific gene expression data to weight or define the intralayer edges, creating state-attributed gene networks.
- Graph Embedding: Generate contextualized embeddings for each gene in each state using a Graph Attention Network (GAT). This step captures both local and global topological features.
- Cross-State Alignment & Shift Quantification: Leverage Gromov-Wasserstein optimal transport to compute the structural shift of each gene's role between states in the learned embedding space.
- Biomarker Ranking: Rank genes with significant alignment shifts using a Dynamic Network Index (DNI), which aggregates scores within connected subnetworks to prioritize biomarkers with coordinated regulatory changes.
- Classification Validation: Apply the prioritized biomarkers in a deep neural network to validate their power in classifying disease states.
The workflow is visualized below.
Protocol for Identifying a Dynamic Network Biomarker (DNB)
DNB identification requires longitudinal data to capture system dynamics [108].
- Longitudinal Sampling: Collect time-series data from a biological system as it approaches a critical transition (e.g., from a disease model or a progressive human cohort).
- Correlation Dynamics Calculation: For each time window, calculate the correlation network between all measured molecules (e.g., genes). Track how these correlations change over time.
- DNB Module Identification: Identify a group of molecules that, as the system nears the critical point, simultaneously exhibits three properties:
- The correlations (PCC) between molecules within the group sharply increase.
- The correlations between molecules inside and outside the group sharply decrease.
- The standard deviations of molecule abundances within the group sharply increase.
- Critical State Warning: The emergence of a module satisfying these conditions serves as a DNB signal, indicating the system is in a pre-disease state and a critical transition is imminent.
The following table details key computational tools and data resources essential for research into network and dynamic biomarkers.
Table 3: Key Research Reagents and Computational Solutions
| Item Name | Type | Primary Function in Research | Example Use Case |
|---|---|---|---|
| Prior Interaction Databases (e.g., STRING, SIGNOR) | Data Resource | Provides prior knowledge of molecular interactions (PPIs, signaling) for network construction [10]. | Used in TransMarker's first step to build the foundational gene network for each disease state [84]. |
| Graph Attention Network (GAT) | Algorithm/Software | A neural network architecture that learns node embeddings by assigning different importance to a node's neighbors [84]. | Generates contextualized, state-specific representations of genes in a network in TransMarker [84]. |
| Optimal Transport (Gromov-Wasserstein) | Mathematical Framework | Computes the structural discrepancy between two networks or their embeddings, aligning them to quantify shifts [84]. | Quantifies the structural rewiring of a gene's regulatory role across different disease states in TransMarker [84]. |
| Cytoscape | Software Platform | An open-source platform for complex network visualization and analysis [10]. | Used to visualize and explore the final network biomarker, identifying key hubs and modules. |
| Single-Cell RNA-Seq Data | Data Type | Provides high-resolution expression profiles at the individual cell level, revealing heterogeneity. | The primary input for the TransMarker framework to study state transitions in cancer [84]. |
| MarkerPredict | Software Tool | A machine learning tool (Random Forest/XGBoost) that integrates network motifs and protein disorder to predict biomarkers [27]. | Identifies potential predictive biomarkers for targeted cancer therapies by analyzing signaling networks. |
The transition from single-molecule markers to network and dynamic network biomarkers represents a fundamental shift from a reductionist to a systems-level understanding of disease. While single-molecule markers remain useful for specific, well-defined conditions, their limitations in specificity and robustness are evident in complex diseases. Network biomarkers offer a more stable and insightful reflection of pathological states by capturing the interplay between molecular components. Dynamic network biomarkers push the frontier further by offering the potential for true prediction, identifying system instability before a drastic transition occurs. The choice of approach depends on the clinical or research question: single markers for simplicity and cost in stable contexts, network biomarkers for nuanced stratification and mechanism, and DNBs for forecasting critical transitions in preventative medicine. As systems biology continues to mature, the integration of these multi-scale biomarkers will be crucial for advancing personalized and predictive healthcare.
For decades, the reductionist approach has dominated drug discovery, operating on the core paradigm that modulating a single gene product can trigger a therapeutic response, and that compounds active against recombinant proteins in vitro will perform similarly in vivo [59]. This "one target, one drug" model has been facilitated by advances in combinatorial chemistry, robotics, and molecular biology [59]. However, despite legitimate expectations that this approach would increase drug discovery frequency while reducing costs, the opposite has occurred—frequency of new drug discovery has decreased while associated costs have surged [59]. The pharmaceutical industry now faces an unacceptable lack of new treatments to address unmet medical needs, particularly for complex diseases in cardiovascular, metabolic, and central nervous system disorders [59].
In response to these limitations, systems biology has emerged as a transformative paradigm that applies computational and mathematical methods to study complex interactions within biological systems [1]. This interdisciplinary field at the intersection of biology, computation, and technology leverages omics datasets to investigate biology as an integrated network rather than as isolated components [1]. Rather than dividing complex problems into smaller units, the systems perspective appreciates holistic and composite characteristics, recognizing that "the forest cannot be explained by studying the trees individually" [109]. This review provides a comprehensive economic and performance comparison between these competing approaches, examining their impacts on drug development efficiency, costs, and success rates.
Methodological Foundations: Core Principles and Experimental Applications
Reductionist Approach: Framework and Limitations
The reductionist drug discovery framework follows a linear pathway beginning with target identification of a single gene product, typically employing biochemical assays using recombinant proteins [59]. This is followed by high-throughput screening of compound libraries against this isolated target, lead optimization focused primarily on target affinity and specificity, and preclinical testing in simplified model systems [59]. The fundamental assumption is that disease pathology can be reversed by modulating a single critical node in biological networks.
Experimental protocols in reductionist approaches typically involve:
- Target Identification: Utilization of genetic association studies, knock-out models, and biochemical studies to identify potential drug targets [59]
- High-Throughput Screening (HTS): Implementation of automated robotic systems to screen hundreds of thousands of compounds against isolated target proteins in biochemical assays [110]
- Lead Optimization: Iterative chemical modification using structure-activity relationship (SAR) analysis to improve potency and selectivity against the single target [110]
- Preclinical Validation: Testing in animal models, often transgenic, that overexpress or lack the target of interest [59]
A critical limitation of this approach is its failure to account for polypharmacology—the fact that most effective drugs interact with multiple targets—and the complex network biology underlying most chronic diseases [59] [111]. Retrospective analysis of approved drugs reveals that the vast majority did not originate from initial primary screening with in vitro assays against single targets, except in rare cases such as anti-infectives [59].
Systems Approach: Integrative Methodologies
Systems biology employs an integrated, holistic framework that begins with comprehensive characterization of disease mechanisms (MOD) through multi-omics data integration [1]. This is followed by network analysis to identify critical pathways and nodes, design of interventions that modulate multiple network components, and validation in complex human cell-based model systems that better recapitulate human physiology [1] [112].
Key experimental methodologies in systems biology include:
- Multi-Omics Data Integration: Simultaneous analysis of genomics, transcriptomics, proteomics, and metabolomics data to construct comprehensive network models of disease biology [1]
- Computational Modeling: Development of mathematical models that simulate network behavior and predict intervention outcomes [1] [112]
- Complex Cell Systems: Utilization of complex human primary cell-based assay systems (e.g., BioMAP systems) that capture multiple pathways and cell types relevant to human disease [113]
- Quantitative Systems Pharmacology (QSP): Implementation of computational platforms that integrate drug properties with system-level biology to predict efficacy and toxicity [112]
This approach explicitly acknowledges that biological systems exhibit emergent properties that cannot be predicted by studying individual components in isolation [109]. It focuses on identifying patterns of response across multiple pathways rather than optimization of single target activity [113].
The diagram below illustrates the fundamental differences in the conceptual frameworks and workflows between reductionist and systems approaches in drug development:
Economic Analysis: Development Costs and Success Rates
Comprehensive Cost Comparison
Drug development costs vary significantly depending on the approach, therapeutic area, and specific development challenges. Recent analyses provide insights into the financial implications of different strategies.
Table 1: Comparative Analysis of Drug Development Costs
| Cost Component | Reductionist Approach | Systems Approach | Data Sources |
|---|---|---|---|
| Direct R&D Cost per Approved Drug | Mean: $369M, Median: $150M [114] | Emerging data suggests potential reduction through improved success rates | RAND study of 38 FDA-approved drugs |
| Full Capitalized Cost (including failures) | Mean: $1.3B, Median: $708M [114] | Projected lower due to earlier failure of unpromising candidates | Analysis accounting for attrition rates |
| Clinical Trial Costs | 60-70% of total R&D budget [110] | Potential reduction through better patient stratification | Industry cost analyses |
| Attrition Rates | >95% failure rate from preclinical to approval [59] | Early detection of failures reduces late-stage costs | Retrospective drug approval studies |
| Cost Drivers | High late-stage failures, poor target validation [59] | Higher initial investment in omics and computational infrastructure | Industry assessments |
The distribution of development costs reveals that a small number of ultra-costly medications skew average development costs, with the mean cost significantly higher than the median cost across recently approved drugs [114]. This suggests that development approaches that reduce outliers could substantially impact overall industry economics.
Success Rates and Attrition Patterns
The most significant economic advantage of systems approaches lies in their potential to improve success rates, particularly in late-stage development where costs are highest. Historical analysis reveals that for complex diseases, "there is not a single instance in the history of drug discovery, where a compound, initially selected by means of a biochemical assay, achieved a significant therapeutic response" [59]. This striking finding underscores the fundamental limitation of reductionist approaches for multifactorial diseases.
Analysis of approved drugs shows that the vast majority exhibit polypharmacology—they achieve their therapeutic effects by acting on multiple gene products rather than single targets [59]. This explains why programs that begin with comprehensive understanding of disease mechanisms and molecular pathways have historically been more successful than those based solely on single-target in vitro screening [59].
Systems approaches address this limitation through:
- Earlier Detection of Failures: Identification of problematic compounds before expensive late-stage development [1]
- Better Target Validation: Understanding targets in their physiological context rather than isolation [113]
- Improved Biomarker Strategies: Patient stratification biomarkers that increase probability of technical success [1] [112]
- Polypharmacology Optimization: Intentional design of multi-target therapies rather than accidental off-target effects [111]
Performance Comparison: Efficacy, Safety, and Development Efficiency
Therapeutic Performance Across Disease Areas
The performance of reductionist versus systems approaches varies significantly across therapeutic areas, with particularly stark differences in complex chronic diseases compared to single-etiology conditions.
Table 2: Therapeutic Performance Comparison by Disease Category
| Disease Category | Reductionist Approach Performance | Systems Approach Performance | Key Differentiators |
|---|---|---|---|
| Infectious Diseases | Strong performance for antibiotics, antivirals [59] | Complementary for host-pathogen interactions | Single pathogen targets often sufficient |
| Oncology | Limited success for most solid tumors | Improved outcomes through combination therapies and biomarkers | Tumor heterogeneity requires multi-target approaches |
| CNS Disorders | Poor track record, high failure rates [59] | Emerging success through network pharmacology | Complex network pathophysiology |
| Cardiovascular & Metabolic | Declining productivity despite investment [59] | Potential for multi-scale modeling of system pathways | Multifactorial pathophysiology |
| Rare Genetic Diseases | Variable depending on monogenic vs complex | Powerful for understanding phenotypic variability | Even monogenic diseases show complex network adaptations |
The performance advantage of systems approaches is most evident in complex diseases where multiple pathways contribute to pathology. For these conditions, single-target modulation often proves insufficient to reverse disease processes, or leads to compensatory mechanisms that diminish therapeutic effects [109] [1].
Development Efficiency and Timeline Analysis
Systems approaches impact not only success rates but also development efficiency through improved decision-making and resource allocation.
The following diagram illustrates how systems approaches integrate multiple data types and computational modeling to enhance decision-making across the development pipeline:
Key efficiency advantages of systems approaches include:
- Reduced Late-Stage Failures: By better predicting human responses earlier in development, systems approaches minimize costly Phase III failures [1]
- Optimized Resource Allocation: Computational models enable prioritization of candidates with higher probability of success [112]
- Accelerated Decision-Making: High-content data from complex cell systems provides more predictive information sooner [113]
- Biomarker-Driven Trials: Patient stratification biomarkers increase trial efficiency and likelihood of success [1]
Research Toolkit: Essential Reagents and Technologies
Key Research Solutions for Implementation
Successful implementation of systems biology approaches requires specialized reagents, technologies, and computational resources.
Table 3: Essential Research Toolkit for Systems Biology in Drug Development
| Tool Category | Specific Solutions | Research Application | Implementation Role |
|---|---|---|---|
| Multi-Omics Platforms | Genomics, transcriptomics, proteomics, metabolomics technologies | Comprehensive molecular profiling of disease states | Characterize mechanism of disease (MOD) and drug effects |
| Complex Cell Systems | Primary human cell co-cultures, 3D organoids, BioMAP platforms | Disease modeling in physiologically relevant contexts | Assessment of compound efficacy and toxicity in human systems |
| Computational Modeling Tools | Quantitative Systems Pharmacology (QSP), PBPK modeling, network analysis | Prediction of drug behavior and system responses | Prioritize candidates, optimize doses, predict clinical outcomes |
| Pathway Analysis Resources | KEGG, Reactome, GeneOntology, custom pathway maps | Biological context for target and drug actions | Identify critical nodes and pathways for therapeutic intervention |
| Data Integration Platforms | Machine learning algorithms, semantic knowledge bases | Integration of diverse data types for pattern recognition | Identify biomarker signatures and drug-pathway associations |
These tools enable researchers to move beyond single-target thinking to network-level interventions. For instance, computational workflows can provide "a boost to accrue big data, with semi-automated and efficient analysis to identify potential drug molecules that can reverse components of the disease mechanistic pathway" [112].
The economic and performance evidence strongly supports a strategic shift toward systems approaches in drug development, particularly for complex diseases. The reductionist paradigm, while successful for single-etiology conditions, has demonstrated fundamental limitations for multifactorial chronic diseases that represent the greatest unmet medical needs and healthcare burdens [59].
The economic case for systems biology rests on its potential to reduce late-stage attrition—the primary driver of development costs—through better target validation, improved biomarker strategies, and more predictive preclinical models [1] [113]. While systems approaches require greater initial investment in technologies and expertise, this upfront cost is likely offset by significant savings from avoided late-stage failures and more efficient resource allocation.
For research organizations, the transition from reductionist to systems approaches represents both a challenge and an opportunity. It requires development of new capabilities in computational biology, data science, and complex cell system modeling [1] [112]. However, organizations that successfully make this transition stand to gain significant competitive advantages through improved development productivity and better alignment with the network pharmacology that underpins most effective medicines [59] [111].
As the field evolves, the most productive path forward likely involves integrating the best aspects of both approaches—the rigorous molecular characterization of reductionism with the network-level understanding of systems biology. This integrated approach promises to address the critical medical needs that have remained elusive under the dominant reductionist paradigm of the past two decades.
The fundamental dichotomy between reductionist and integrative systems approaches represents a critical philosophical divide in contemporary biological research, particularly in the field of biomarker discovery and drug development. The reductionist approach, which has dominated biomedical science for decades, operates on the principle that complex systems can be understood by breaking them down into their constituent parts and studying each component in isolation [115]. This framework aligns with Francis Crick's 'Central Dogma of Molecular Biology,' which posits a linear flow of genetic information from DNA to RNA to protein [116]. While this paradigm has yielded tremendous insights into molecular mechanisms, its limitations are increasingly apparent when addressing complex biological phenomena where emergence, interactions, and network dynamics play decisive roles [115].
In contrast, integrative systems biology represents a philosophical shift toward understanding biological systems as interconnected networks rather than collections of isolated components [117]. As articulated by Dennis Noble, "Systems biology...is about putting together rather than taking apart, integration rather than reduction" [115]. This approach acknowledges that "the whole becomes not merely more, but very different from the sum of its parts" [115], recognizing that emergent properties arise from complex interactions that cannot be predicted by studying individual components alone. The paradigm conflict between these approaches has profound implications for biomarker discovery, therapeutic development, and our fundamental understanding of disease mechanisms.
Performance Comparison: Quantitative Outcomes Across Methodologies
Table 1: Comparative Analysis of Research Outcomes Between Approaches
| Performance Metric | Reductionist Approach | Integrative Systems Approach | Evidence Source |
|---|---|---|---|
| Hub Genes Identified | Single candidate biomarkers | 99 central hub genes identified in colorectal cancer study [33] | Colorectal Cancer Network Analysis |
| Diagnostic Biomarker Efficiency | CCNA2, CD44, ACAN individually associated with poor prognosis [33] | Combined biomarker panels with network centrality | Colorectal Cancer Study |
| Survival Association Signals | Limited to pre-selected targets | TUBA8, AMPD3, TRPC1, ARHGAP6, JPH3, DYRK1A, ACTA1 associated with decreased survival [33] | Survival Analysis Validation |
| Therapeutic Target Discovery | Single pathway targets | MMP9, POSTN, HES5 identified as key nodes with existing drug associations [118] | Glioblastoma Multiforme Study |
| Network Context | Limited or no network context | 7 interactive modules with functional specialization [33] | Module Identification |
Table 2: Experimental Validation Outcomes in Disease Models
| Disease Context | Systems Biology Discovery | Experimental Validation Outcome | Therapeutic Impact |
|---|---|---|---|
| Lung Cancer (TGF-β/EMT) | ATG16L1 identified as central node in amine metabolism network [119] | siRNA knockdown re-sensitized cells to therapies [119] | Overcame chemoresistance |
| Glioblastoma Multiforme | MMP9 with highest degree in hub biomarker network [118] | Molecular docking confirmed high binding affinities (-6.3 to -8.7 kcal/mol) [118] | Identified carmustine, marimastat as potential therapeutics |
| Colorectal Cancer | 99 hub genes through centrality analysis [33] | Survival analysis confirmed prognostic value [33] | Multiple biomarker and target candidates |
Integrative frameworks demonstrate superior performance across multiple quantitative metrics, particularly in the comprehensiveness of biomarker identification and functional context provided. Where reductionist methods might identify individual candidates, systems approaches reveal entire interactive networks. In colorectal cancer research, the integrative approach identified 99 hub genes through protein-protein interaction (PPI) network analysis compared to the handful typically discovered through reductionist methods [33]. More importantly, these genes were contextualized within seven interactive modules with distinct functional specializations, providing not just biomarkers but functional pathways for therapeutic intervention.
The therapeutic implications are equally significant. In lung cancer research focusing on TGF-β-mediated epithelial-mesenchymal transition (EMT), phylogenetic clustering of gene expression data revealed convergence toward amine metabolic pathways and autophagy [119]. This systems-level insight led to the experimental validation that ATG16L1 knockdown re-sensitized resistant cancer cells to therapies—a finding that emerged from understanding network dynamics rather than isolated components [119]. Similarly, glioblastoma research identified MMP9 as the highest-degree node in hub biomarker networks, with molecular docking confirming strong binding affinities for existing drugs, potentially repurposing them for this aggressive cancer [118].
Methodological Comparison: Experimental Protocols and Workflows
Reductionist Approach Protocol
Objective: Isolate and characterize individual biomarker candidates in disease processes.
Methodology:
- Hypothesis-Driven Investigation: Begin with pre-defined candidate biomarkers based on existing literature
- Targeted Assays: Utilize focused measurement techniques (Western blot, ELISA, qPCR) for specific molecules
- Linear Causality Modeling: Assume straightforward cause-effect relationships
- Single-Variable Optimization: Control all variables while testing one specific factor
- Validation in Simplified Models: Confirm findings in controlled cell culture or animal models
Limitations: This approach "overlooks and thus cannot prognosticate on the formidable unintended consequences that emerge from 'doing the right things wrong'" [120] and fails to account for network effects and emergent properties that characterize complex biological systems [115].
Integrative Systems Biology Protocol
Objective: Understand disease mechanisms through comprehensive network analysis and identify robust biomarker panels.
Methodology:
- Data Acquisition: Collect multi-omics data (genomics, transcriptomics, proteomics) from appropriate samples (e.g., GEO dataset GSE11100 for glioblastoma [118])
- Differential Expression Analysis: Identify statistically significant DEGs using R/Bioconductor packages with p-values and false discovery rates [118]
- Network Construction: Reconstruct protein-protein interaction (PPI) networks using STRING database and visualize with Cytoscape [33] [118]
- Centrality Analysis: Identify hub genes based on network topology metrics (degree, betweenness, closeness) [33]
- Module Detection: Apply clustering algorithms (k-means) to identify functional modules [33]
- Enrichment Analysis: Annotate biological functions and pathways using Gene Ontology (GO) and KEGG databases [118]
- Survival Analysis: Validate clinical relevance using tools like GEPIA [33]
- Experimental Validation: Confirm computational predictions through in vitro and in vivo models [119]
Workflow Visualization: Integrative Systems Biology Approach
Table 3: Core Reagents and Computational Tools for Integrative Systems Biology
| Category | Specific Tools/Reagents | Function/Purpose | Application Example |
|---|---|---|---|
| Data Sources | GEO Database [118], STRING [33] | Repository for gene expression data; Protein-protein interaction networks | Retrieval of GSE11100 for glioblastoma study [118] |
| Analysis Software | Cytoscape [119] [33], Gephi [33] | Network visualization and analysis; Network visualization and centrality analysis | PPI network reconstruction and hub identification [33] |
| Bioconductor Packages | R/Bioconductor [33] | Differential gene expression analysis | Identification of DEGs with statistical significance [33] |
| Validation Tools | GEPIA [33], Molecular docking software [118] | Survival analysis; Binding affinity prediction | Prognostic value assessment of hub genes [33] |
| Experimental Reagents | siRNA for ATG16L1 [119] | Gene knockdown to validate target function | Resensitization of lung cancer cells to therapies [119] |
Signaling Pathways and Network Dynamics in Disease
The integrative approach reveals that diseases often converge on specific signaling pathways through evolutionary processes. In lung cancer research, phylogenetic analysis of gene expression data during TGF-β-mediated EMT revealed convergence toward amine metabolic pathways and autophagy regulation [119]. This convergence suggests these pathways represent critical vulnerabilities in therapy-resistant cancers.
TGF-β/EMT Signaling Network
This network visualization illustrates how the integrative approach maps the connections between initial signaling events (TGF-β activation), intermediate processes (EMT, metabolic reprogramming), and ultimately phenotypic outcomes (chemoresistance). The identification of ATG16L1 as a key node connecting autophagy to chemoresistance emerged from this systems-level analysis [119], demonstrating how integrative frameworks reveal non-obvious connections that might be missed in reductionist studies.
Comparative Strengths and Limitations in Research Applications
Table 4: Framework Performance Across Research Applications
| Research Application | Reductionist Advantages | Integrative Framework Advantages |
|---|---|---|
| Biomarker Discovery | Rapid validation of individual candidates | Comprehensive biomarker panels with built-in validation through network properties [33] |
| Drug Target Identification | Straightforward mechanistic studies | Identification of central nodes in disease networks with higher therapeutic potential [118] |
| Understanding Resistance | Focused on specific resistance mechanisms | Reveals network-level adaptations and convergent evolution toward vulnerable pathways [119] |
| Predictive Modeling | Simple linear models | Incorporates emergent properties and feedback loops for more accurate predictions [115] |
| Clinical Translation | Simplified diagnostic development | Multi-biomarker signatures with potentially higher specificity and sensitivity [33] |
The integrative framework demonstrates particular strength in addressing complex diseases like cancer, where robustness and adaptive capacity emerge from network properties rather than individual components. As noted in critical assessments of the reductionist approach, "the extreme reductionist approach and heavy reliance on the so-called molecular biology in recent years has become a negative factor and has occluded the enormously exciting view that biology presents today" [117]. The ability to map and understand network-level adaptations provides explanatory power for phenomena like therapeutic resistance that often frustrate reductionist approaches.
The evidence synthesized across multiple disease contexts reveals both quantitative and qualitative advantages of integrative frameworks over strictly reductionist approaches. Integrative systems biology provides more comprehensive biomarker panels, reveals functional modules within disease networks, identifies non-obvious therapeutic targets, and ultimately offers more robust predictive models of complex biological behavior.
Rather than representing mutually exclusive paradigms, these approaches can be complementary. Reductionist methods provide crucial mechanistic insights and validation, while integrative frameworks provide the essential context for understanding system-level behaviors [120]. The future of biomarker discovery and therapeutic development lies in leveraging the strengths of both approaches—using integrative methods to identify key nodes and networks, followed by reductionist approaches to elucidate detailed mechanisms.
This synthesis suggests that research institutions and funding agencies should prioritize approaches that combine high-throughput data generation with sophisticated computational analysis and experimental validation. The most promising path forward involves iterative cycles of computational model building and experimental refinement [115] [119], leveraging the power of both reductionist and integrative thinking to advance our understanding and treatment of complex diseases.
Conclusion
The integration of systems biology into biomarker research represents a fundamental evolution beyond reductionist approaches, enabling a more comprehensive understanding of complex diseases through multi-omics integration, computational modeling, and network analysis. This paradigm shift addresses critical limitations of single-target hypotheses by capturing the dynamic interactions within biological systems, leading to more robust biomarkers, improved patient stratification, and enhanced therapeutic development. The synergistic combination of systems biology with artificial intelligence is particularly powerful, creating an 'Iterative Circle of Refined Clinical Translation' that continuously improves both products and clinical strategies. Future directions will focus on standardizing analytical frameworks, enhancing computational models for better clinical predictability, and fully realizing personalized, predictive, and preventive medicine. For researchers and drug developers, adopting these integrative approaches is becoming increasingly essential for tackling the most pressing challenges in modern biomedicine and delivering effective, patient-centric therapies.
References
- 1. Systems biology platform for efficient development and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12341954/]
- 2. Machine Learning in Biomarker Discovery for Precision ... [https://esmed.org/machine-learning-in-biomarker-discovery-for-precision-medicine/]
- 3. Emerging Technologies in Biomarker Discovery You ... [https://blog.crownbio.com/emerging-technologies-in-biomarker-discovery-you-should-know-about]
- 4. Quantifying and managing uncertainty in systems biology [https://www.sciencedirect.com/science/article/pii/S2452310025000174]
- 5. Disclosure of Alzheimer's disease blood-based biomarker ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12501330/]
- 6. Insights from Biomarkers & Precision Medicine 2025 [https://www.signifyresearch.net/insights/biomarkers-billions-and-breakthroughs-insights-from-biomarkers-precision-medicine-2025/]
- 7. AI and the future of biomarker analysis in early R&D [https://www.drugtargetreview.com/article/190428/ai-and-the-future-of-biomarker-analysis-in-early-rd/]
- 8. Applications and challenges of biomarker-based predictive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12399543/]
- 9. AI-Powered Biomarker Discovery: 7 ... [https://lifebit.ai/blog/ai-powered-biomarker-discovery-2/]
- 10. Systems Biology as a Comparative Approach to ... [https://www.mdpi.com/2076-328X/3/2/253]
- 11. Systems biology approach highlights mechanistic ... [https://www.nature.com/articles/s41598-021-91124-3]
- 12. Biomarker discovery pipeline: 3 Critical Steps 2025 [https://lifebit.ai/blog/biomarker-discovery-pipeline/]
- 13. Challenges and Future Trends in Biomarker Analysis for ... [https://blog.crownbio.com/challenges-and-future-trends-in-biomarker-analysis-for-clinical-research-whats-ahead-in-2025]
- 14. A Brief History of Systems Biology - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC1626627/]
- 15. Systems biology [https://en.wikipedia.org/wiki/Systems_biology]
- 16. Systems Biology Integration → Term [https://climate.sustainability-directory.com/term/systems-biology-integration/]
- 17. Methodological developments in systems biology [https://www.nature.com/collections/chdbdhfibd]
- 18. The Role of Systems Biology in Therapeutic Innovation [https://www.walshmedicalmedia.com/open-access/the-role-of-systems-biology-in-therapeutic-innovation-135216.html]
- 19. Expanding our thought horizons in systems biology and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12341958/]
- 20. Bridging academia and industry: advancing systems ... [https://www.frontiersin.org/journals/systems-biology/articles/10.3389/fsysb.2025.1627214/full]
- 21. Causal thinking and complex system approaches in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2912489/]
- 22. Time to change from a simple linear model to a complex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4886826/]
- 23. Interactome Networks and Human Disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3102045/]
- 24. Pursuit of precision medicine: Systems biology approaches in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10112395/]
- 25. Network-based approaches for modeling disease ... [https://www.sciencedirect.com/science/article/pii/S2001037022005797]
- 26. Identification of disease treatment mechanisms through the ... [https://www.nature.com/articles/s41467-021-21770-8]
- 27. MarkerPredict: predicting clinically relevant ... [https://www.nature.com/articles/s41540-025-00603-0]
- 28. Challenges and opportunities in the network medicine of ... [https://www.sciencedirect.com/science/article/pii/S2666634025003472]
- 29. A quantum mechanics-based framework for infectious ... [https://www.nature.com/articles/s41598-025-96817-7]
- 30. Systems Biology as a Comparative Approach to Understand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4217627/]
- 31. A guide to multi-omics [https://frontlinegenomics.com/a-guide-to-multi-omics/]
- 32. The Power of High-Throughput Omics Integration - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417901/]
- 33. Systems biology approach to identify biomarkers and ... [https://www.sciencedirect.com/science/article/pii/S2405580823002145]
- 34. Systems biology platform for efficient development and ... [https://www.frontiersin.org/journals/systems-biology/articles/10.3389/fsysb.2023.1229532/full]
- 35. Systems biology approaches to identify driver genes and ... [https://www.nature.com/articles/s41598-024-52484-8]
- 36. High performance data integration for large-scale analyses ... [https://www.nature.com/articles/s41467-025-62237-4]
- 37. Visualization of three-way comparisons of omics data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1831488/]
- 38. Emerging technologies of single-cell multi-omics - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/39911109/]
- 39. livermetabolism [http://www.livermetabolism.com/]
- 40. Programmatic modeling for biological systems [https://www.sciencedirect.com/science/article/pii/S2452310021000287]
- 41. A Review on Biomarker‐Enhanced Machine Learning for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12421415/]
- 42. The Impact of Pathway Database Choice on Statistical ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2019.01203/full]
- 43. A systematic comparison of the MetaCyc and KEGG pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3665663/]
- 44. What are the advantages and disadvantages between ... [https://bioinformatics.stackexchange.com/questions/230/what-are-the-advantages-and-disadvantages-between-using-kegg-or-reactome]
- 45. Cobra Toolbox and Cobrapy [https://systemsbiology.ucsd.edu/Downloads/CobraToolbox]
- 46. Converters [https://sbml.org/software/converters/]
- 47. Systems Biology Markup Language (SBML) Level 3 Package [https://pmc.ncbi.nlm.nih.gov/articles/PMC7756622/]
- 48. Systems Medicine: The Application of Systems Biology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4556074/]
- 49. Application of Top-Down and Bottom-up Systems Approaches ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3401895/]
- 50. Toward merging bottom–up and top–down model-based ... [https://www.sciencedirect.com/science/article/pii/S1369527422000534]
- 51. Which is Best for Us? Top Down, Bottoms Up, or Middle Out [https://www.bpminstitute.org/resources/articles/which-best-us-top-down-bottoms-or-middle-out/?srsltid=AfmBOoreiiYfNyql5rzbaZGndBYJVyPHutXSTPPWtSvW8vKdpL5PgDB0]
- 52. Top-down, Middle-out, and Bottom-up - systems-wise.com [https://systems-wise.com/the-three-systems-engineering-approaches-top-down-middle-out-and-bottom-up/]
- 53. Top-Down versus Bottom-Up Approaches in Proteomics [https://www.chromatographyonline.com/view/top-down-versus-bottom-approaches-proteomics-0]
- 54. Top Down, Middle Out, Bottom Up: What's Right For Me? [https://stockiqtech.com/blog/top-down-middle-out-bottom-up/]
- 55. The heterogeneity of mesenchymal stem cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC10734083/]
- 56. The issue of heterogeneity of MSC-based advanced ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1400347/full]
- 57. Challenges and advances in clinical applications of ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-021-01037-x]
- 58. Cause and consequence of heterogeneity in human ... [https://www.sciencedirect.com/science/article/abs/pii/S0344033824002656]
- 59. The inadequacy of the reductionist approach in discovering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6180410/]
- 60. Artificial intelligence and systems biology analysis in stem cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12476622/]
- 61. Based Systems Biology Approaches in Multi-Omics Data ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2020.588221/full]
- 62. Precision neurodiversity: personalized brain network ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12647089/]
- 63. Quantitative Data Analysis. A Complete Guide [2025] [https://www.6sigma.us/six-sigma-in-focus/quantitative-data-analysis/]
- 64. Review article Biomarkers: Promising and valuable tools ... [https://www.sciencedirect.com/science/article/pii/S2405844023005303]
- 65. Integrating Multi-Omics Data: Methods and Applications in ... [https://www.sciencedirect.com/science/article/pii/S2215017X25000657]
- 66. Bridging Discovery and Treatment: Cancer Biomarker - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12651302/]
- 67. Gene- and immune-targeted therapy combinations using ... [https://www.nature.com/articles/s41698-025-01038-w]
- 68. The latest trends in drug discovery [https://www.cas.org/resources/cas-insights/2025-drug-discovery-trends]
- 69. Balancing ethical data sharing and open science for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12047515/]
- 70. The Biggest AI Challenge in Biomedical Research [https://www.snapcyte.com/ai-data-bottleneck/]
- 71. Data- and AI-intensive Research with Rigor and ... - MIDAS [https://midas.umich.edu/events/dair-sa-2025/]
- 72. Integrative research: Current trends and considerations for ... [https://www.sciencedirect.com/science/article/pii/S2950194625001360]
- 73. Advancements in multi-omics research to address ... [https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2025.1591796/full]
- 74. Machine learning comparison for biomarker level ... [https://www.nature.com/articles/s41598-025-16185-0]
- 75. Seeking optimal repeated fluid biomarker assessments to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12541280/]
- 76. What Are Biomarkers? Definition, Applications and ... [https://blog.omni-inc.com/blog/what-are-biomarkers-and-whats-new-in-2025]
- 77. Systems biology and the discovery of diagnostic biomarkers [https://pmc.ncbi.nlm.nih.gov/articles/mid/NIHMS259366/]
- 78. Systems biology-based analysis exploring shared ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1398990/full]
- 79. AI in drug discovery: Key insights from a computational ... [https://www.digital-science.com/blog/2025/10/ai-in-drug-discovery-key-insights/]
- 80. A systems biology approach to dynamic modeling and inter ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3117731/]
- 81. Modular assembly of dynamic models in systems biology [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009513]
- 82. Systems biology of cancer biomarker detection - PubMed [https://pubmed.ncbi.nlm.nih.gov/24240581/]
- 83. Bridging the Gap : Translating Preclinical Biomarkers to Clinical Success [https://blog.crownbio.com/bridging-the-gap-translating-preclinical-biomarkers-to-clinical-success]
- 84. TransMarker: Unveiling dynamic network in cancer... biomarkers [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013743]
- 85. Neutrophil extracellular trapping network -associated biomarkers in... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06819-2]
- 86. Unraveling patient heterogeneity in complex diseases through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10449456/]
- 87. Distinct genetic liability profiles define clinically relevant ... [https://www.nature.com/articles/s41467-024-49338-2]
- 88. A data-driven, knowledge-based approach to biomarker ... [https://www.nature.com/articles/s41540-018-0056-1]
- 89. Use of Biomarkers in Drug Development for Regulatory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12489174/]
- 90. Preclinical vs. Clinical Biomarkers: Understanding Their ... [https://blog.crownbio.com/preclinical-vs-clinical-biomarkers-understanding-their-distinct-roles-in-drug-development]
- 91. DRIVe | Type to Treat | Therapeutic Efficacy through Predicting ... [https://drive.hhs.gov/typetotreat.html]
- 92. How Biomarkers Can Improve the Drug Development ... [https://www.fda.gov/drugs/biomarker-qualification-program/biomarker-qualification-program-educational-module-series-module-2-how-biomarkers-can-improve-drug]
- 93. Reductionism Gives Way to Systems Biology [https://www.genengnews.com/insights/reductionism-gives-way-to-systems-biology/]
- 94. Training Interdisciplinary Scientists for Systems Biology [https://pmc.ncbi.nlm.nih.gov/articles/PMC9886957/]
- 95. Systems immunology: When systems biology meets immunology [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447193/]
- 96. Systems biology approaches identify metabolic signatures ... [https://www.nature.com/articles/s41467-024-52909-y]
- 97. Barriers to Interdisciplinary Research and Training - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK44876/]
- 98. An architecture for collaboration in systems biology at the ... [https://www.nature.com/articles/s41540-024-00334-8]
- 99. Team principles for successful interdisciplinary research ... [https://www.sciencedirect.com/science/article/pii/S2666602223000587]
- 100. Human disease biomarker panels through systems biology [https://pmc.ncbi.nlm.nih.gov/articles/PMC8724340/]
- 101. Assessment of diagnostic and prognostic accuracy - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK260277/]
- 102. Comparative evaluation of feature reduction methods for ... [https://www.nature.com/articles/s41598-024-81866-1]
- 103. A step-by-step guide to the systematic review and meta- ... [https://www.nature.com/articles/bjc2013185]
- 104. Integrated Biomarker Discovery Workflow-Bioinformatics to ... [https://www.elucidata.io/blog/integrated-biomarker-discovery-workflow-bioinformatics-to-clinical-validation]
- 105. Biomarker Discovery and Validation: Statistical Considerations [https://pmc.ncbi.nlm.nih.gov/articles/PMC8012218/]
- 106. A systematic comparison of ATN biomarkers for monitoring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12545699/]
- 107. Clinical Validation of a Machine Learning-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11857289/]
- 108. Molecular biomarkers, network biomarkers, and dynamic ... [https://www.sciencedirect.com/science/article/pii/S2667325822003065]
- 109. The Limits of Reductionism in Medicine: Could Systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1459480/]
- 110. Which part in drug development is more costly ... [https://synapse.patsnap.com/article/which-part-in-drug-development-is-more-costly-drug-discovery-or-clinical-trials]
- 111. Systems Biology: A New Paradigm for Drug Discovery [https://www.sciencedirect.com/science/chapter/edited-volume/pii/B9780124172050000171]
- 112. A leap in to systems biology - Research Communities [https://communities.springernature.com/posts/a-leap-in-to-systems-biology]
- 113. Can cell systems biology rescue drug discovery? [https://www.nature.com/articles/nrd1754]
- 114. Typical Cost of Developing a New Drug Is Skewed by Few ... [https://www.rand.org/news/press/2025/01/07.html]
- 115. Integrative physiology and systems biology: reductionism ... [https://extremephysiolmed.biomedcentral.com/articles/10.1186/2046-7648-2-9]
- 116. Current advances in systems and integrative biology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4212281/]
- 117. Reductionist vs integrative approach in biology [https://indiabioscience.org/columns/education/reductionist-vs-integrative-approach-in-biology]
- 118. System biology approach to identify the novel biomarkers ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1364138/full]
- 119. An Integrative Systems Biology and Experimental Approach ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6406733/]
- 120. A Review of Reductionist versus Systems Perspectives ... [https://www.mdpi.com/2079-8954/9/2/38]